Figures & data
Figure 1. Metformin inhibits RANKL gene expression in breast epithelial cells. Total RNA from untreated and metformin-treated (5 mmol/L; 48 h) BRCA1+/+ and BRCA1mut/+ MCF10A isogenic cell pairs was evaluated in technical triplicates for the abundance of RANK (TNFRSF11A, Hs00921372_m1) and RANKL (TNFSF11, Hs00243522_m1) relative to housekeeping genes GADPH (Hs99999905_m1) and 18S (Hs99999901_s1). The transcript abundance was calculated using the delta Ct method and presented as relative quantification (RQ) or log2 fold-change, as specified. MET, Metformin.
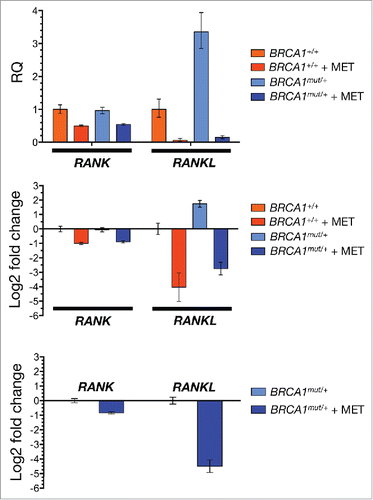
Figure 2. Metformin sensitizes mammosphere-initiating cells to denosumab. Cell2Sphere™ assays using BRCA1-deficient MDA-MB-436 cells were performed as per the manufacturer's instructions ((http://stemtektherapeutics.com/en/cell2sphere#cell2sphere_kit). Drugs were added to quintuplicate sets of wells on days 1 and 4 without replenishing the medium. ImageJ was used to quantify the size (left; central lines indicate mean values) and number (right) of 6-day-old mammospheres. *P < 0.05, **P < 0.001 MET, Metformin. Size bar = 2000 μm.
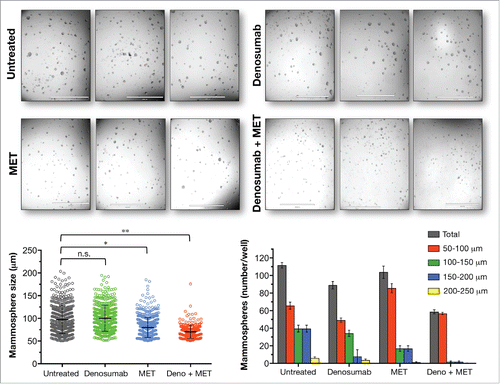
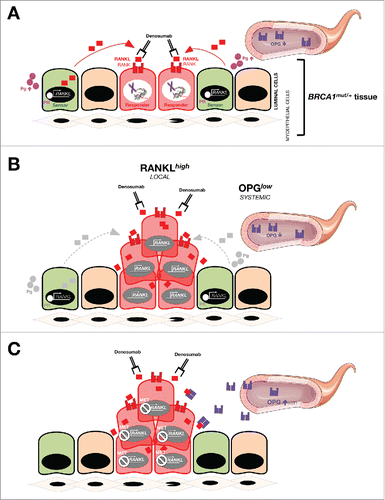
Figure 3. Dysregulation of the OPG/RANKL/RANK signaling axis and BRCA1 deficiency-driven breast oncogenesis: (A)new target for anti-cancer metformin. Molecular dialog between progesterone (Pg)-responsive cells and hormone receptor (HR)-negative luminal progenitors seems to drive the hyperproliferation of breast cancer-initiating cell populations within the breast epithelia of BRCA1 mutation carriers.Citation11,12 In a hormone-dependent stage (e.g., pre-menopausal women), RANKL secretion might become stimulated upon binding of Pg to its receptor (PR) in mature ductal cells (i.e., so-called “sensor” cells); secreted RANKL then binds and activates RANK on RANK+ luminal progenitors (i.e., so-called “responder” cells). Generally, OPG negatively regulates RANKL/RANK signaling by binding to RANKL as an endogenous decoy receptor, thereby inhibiting signal transduction. However, the mitogenic cross-talk between RANKL-producing sensor cells and RANKL-addicted responder cells might be less restricted in high-risk subgroups of BRCA1 mutation carriers with significant lower plasma levels of OPG. The net magnitude of the OPG/RANKL/RANK signaling axis in driving BRCA1-related breast oncogenesis may be largely determined by local increases of RANKL along with decreases in systemic OPG. Our recent findings in HR-negative, basal-like MCF10A breast epithelial cells with a pathogenic185delAG mutation in one BRCA1 alleleCitation13 further suggest that, in the absence of hormone influence, breast cancer-initiating cells may remain responsive to RANKL stimulation because compromised DNA repair and consequent genomic instability provoked by BRCA1 haploinsufficiency might suffice to establish hyperplasic states via an auto-regulatory feedback loop triggered by cell-autonomous hyperactivation of RANKL gene expression. Such a hormone-independent scenario might become reinforced by low circulating levels of OPG not only in post-menopausal BRCA1 carriers but also in other post-menopausal groups at high risk for breast cancer and in women with circulating tumor cells. The ability of metformin (MET) to restore dysregulated OPG/RANKL/RANK signaling may represent an important clinical opportunity to prevent breast cancer and maintain bone health (e.g., following salpingo-oophorectomy) in BRCA mutation carriers and to treat RANKL-driven metastasis-initiating lesions in combination with denosumab.