Figures & data
Figure 1. Imbalanced expression/ activity of RAD18 and Y-family DNA polymerases dictates choice of TLS polymerases and can influence replication fidelity and genome stability. (A) In normal cells TLS polymerases are activated sparingly and used selectively to minimize error-prone DNA synthesis and ensure genome stability. (B) In XPV cells lacking functional Polh, compensatory bypass of Polh-cognate lesions by inappropriate DNA polymerases leads to mutagenesis. (C) In many cancer cells RAD18 is expressed at high levels (sometimes owing to stabilization by its binding partner MAGE-A4), leading to increased TLS pathway activation. It is not known whether all Y-family DNA polymerases are equally dependent on RAD18 for activation. Differential activation of Y-family TLS polymerases by over-active RAD18 would constitute a mechanism of imbalance and altered TLS. (D) Over-expression or reduced expression of individual TLS polymerases has been reported in many cancers and represents another mechanism of TLS pathway imbalance and mutagenesis.
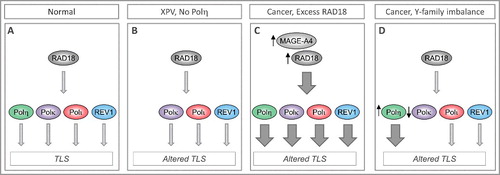
Figure 2. The potential DNA polymerase η mutational signature (Signature 9, http://cancer.sanger.ac.uk/cosmic/signatures). The mutation context (+1 and −1 positions) are shown below frequency bars. This signature (together with more than 20 other distinct mutational signatures) was extracted using a classification analysis of 4,938,362 mutations from 7,042 cancers [Citation26].
![Figure 2. The potential DNA polymerase η mutational signature (Signature 9, http://cancer.sanger.ac.uk/cosmic/signatures). The mutation context (+1 and −1 positions) are shown below frequency bars. This signature (together with more than 20 other distinct mutational signatures) was extracted using a classification analysis of 4,938,362 mutations from 7,042 cancers [Citation26].](/cms/asset/e11d10db-5f8a-43f4-bf8e-2ee9c7e8ee35/kccy_a_1456296_f0002_oc.jpg)
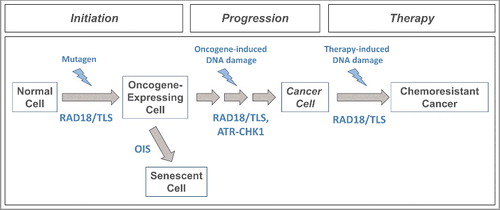
Figure 3. Proposed roles of RAD18 and TLS DNA polymerases in tumorigenesis. Mutagenic TLS of damaged DNA by Y-family DNA polymerases can activate oncogenes (e.g. KRASV12) that initiate carcinogenesis. Oncogene-expressing cells can be eliminated via a DNA damage-mediated senescence program (OIS) that serves as a barrier to tumorigenesis. RAD18/TLS and ATR-CHK1 help sustain damage-tolerant DNA synthesis and viability of cells that breach the OIS barrier. Error-prone TLS might also confer mutability that drives subsequent stages of multi-step tumorigenesis. The selective pressure for cancer cells to activate TLS also confers chemoresistance.