
Figures & data
Figure 1. Effects of age on AD-like pathology with amyloid-beta accumulation. In Sgo1−/+ model, amyloid-beta accumulation in the brain was not observed at the age of 12 months, but was observed at the age of 24 months and older. The accumulation occurred both in the hippocampus and in the cortex, with significant accumulation indicated in the cortex. Various factors that represent “age” have been proposed (see text).
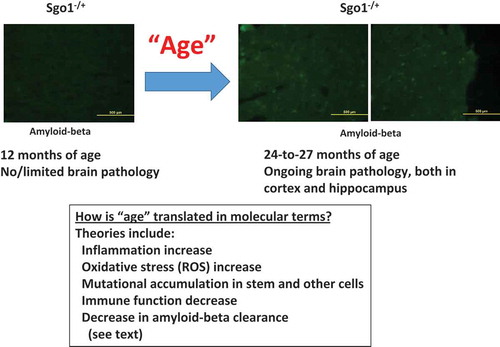
Figure 2 Critical role of prolonged mitosis in amyloid-beta accumulation, suggested by Sgo1−/+ model. (a) Two consequences of Sgo1−/+ with common destinationSgo1−/+ induces two defects in the cell cycle; 1) Mitotic chromosome cohesion defect followed by premature chromosome segregation and chromosome instability, and 2) Centrosome integrity defect followed by aberrant multipolar mitosis. Both defects provoke the mitotic spindle checkpoint that leads to prolonged mitosis. Amyloid-beta (and p-TAU and BACE) accumulation occurred in mitotic marker p-H3-positive, live cells. We propose that the amyloid-beta accumulating cells in prolonged mitosis state eventually die of mitotic catastrophe, releasing amyloid-beta to extracellular matrix. As amyloid-beta possesses prion-like self-aggregating property, the released amyloid-beta may serve as “seeds” for further aggregation and eventual development of neurotoxic senile plaques. (b) “Mitotic origin of deposits” model. Whether amyloid-beta accumulating cells are originated from (case1) terminally differentiated neuron or (case 2) mitotically competent (stem-like) cells has not been determined. In “Hypothetical Case 1: from terminally differentiated neuron”, terminally differentiated neuronal cells de-differentiate to stem-like cells, or receive mitogenic signal and re-enter mitotic cycle. In “Hypothetical Case 2: from mitotically competent (stem-like) cells”, mitotically-competent stem (-like) cells “got stuck in mitosis” possibly due to aneuploidy or other mutation/stimulus that provokes spindle checkpoint leading to mitotic prolongation. In either case, cells in the state of prolonged mitosis are the source of amyloid-beta accumulation. When these cells die (of mitotic catastrophe), the accumulated amyloid-beta is released to extracellular matrix, followed by senile plaque development.
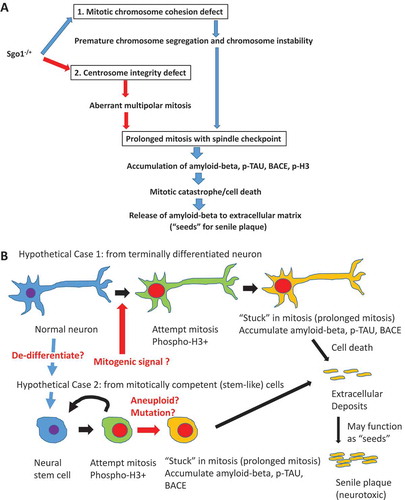
Figure 3. “Injury-wound healing response” model for mitotic re-entry. How mitotic re-entry occur is a key question. Known LOAD risk factors (diabetes, hypertension, stroke) all provoke small or large injuries in the brain. Genomic instability including Sgo1−/+ can also cause cell death, a form of micro-injury. Such injuries activate growth signaling associated with wound-healing (NFkB/JNK, GSK or MAPK). The wound/injury-activated growth signaling may play a significant role in facilitating mitotic re-entry in the brain.
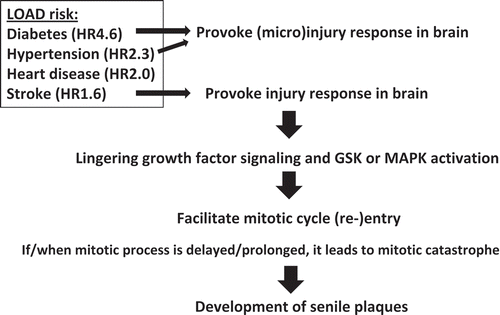
Figure 4. The “three-hit” hypothesis. Based on results from Sgo1−/+ mouse model and existing theories from human LOAD studies, we propose the “three-hit” hypothesis for LOAD. When the “Three-hit” (age, mitotic re-entry, and mitotic prolongation) occurs simultaneously, brain cells accumulate amyloid-beta during prolonged mitosis. Prolonged mitosis may be followed by cell death via mitotic catastrophe, leading to release of amyloid-beta to extracellular matrix (see legend). Various factors lead to each “hit”. “Age” can be translated to a variety of insults (see text). Mitotic re-entry may have been caused by injuries and activation of growth signaling associated with wound/injury healing. Mitotic prolongation is caused by spindle checkpoint activation. Spindle checkpoint activation can occur by conditions commonly found in LOAD brains, such as aneuploidy or cohesinopathy.
