Figures & data
Figure 1. Nutrient-induced regulation of autophagy. Nutrient deprivation and therapy stress activate the metabolic sensors mTOR and AMP-activated kinase (AMPK) which act as major molecular switches of autophagy in response to nutrients. Under conditions of sufficient nutrients, mTOR complex 1 (mTORC1) is activated by upstream regulators, such as amino acid-sensing pathways. Active mTORC1 negatively regulates autophagy induction by phosphorylating and inactivating the ULK1 complex (ULK1/FIP200/ATG13) and the VPS34/BECN1complex (VPS34/BECN1/ATG14/VPS15). When nutrients are limiting, such as under glucose starvation, activated AMPK directly promotes autophagy by phosphorylating and activating the ULK1 complex and the VPS34/BECN1 complex. Activated AMPK and ULK1 negatively regulate mTORC1 activity, thereby relieving its inhibitory effect on autophagy. Autophagy starts from an isolation membrane (phagophore), followed by the formation of a double-membraned autophagosome, which engulfs intra-cellular proteins and subcellular organelles. Autophagosomes then mature by fusion with lysosomes, leading to the degradation of autophagosomal contents. Cells survive in the case of limited autophagy, such as in therapy-resistant cancer cells, whereas with high levels of autophagy cells will succumb to cell death
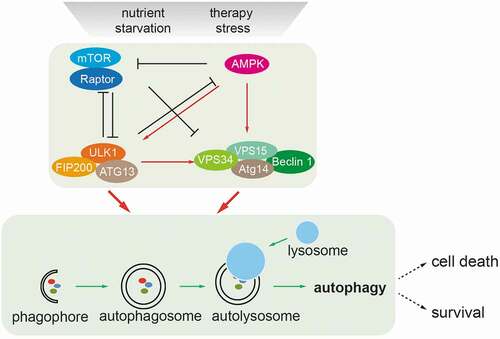
Figure 2. Schematic representation of the mammalian Hippo signaling pathway. Large Tumor Suppressor kinases (LATS1/2) can be activated by a variety of signals, including by mammalian Hippo kinases MST1/2 or other upstream kinases (MAP4Ks), by G-protein coupled receptor (GPCR)-mediated modulation of Rho family GTPase activities and/or actin cytoskeleton remodeling, and by NF2-mediated membrane recruitment. In turn, activated LATS1/2 phosphorylate and inactivate YES-associated protein (YAP) on Ser127 and transcriptional co-activator with PDZ-binding motif (TAZ) on Ser89. YAP/TAZ are transcriptional co-activators that interact with DNA binding transcription factors (TFs), such as TEADs, to initiate gene expression programs. Cell junctional α-catenin (α-CAT) or angiomotin (AMOT) inactivate YAP/TAZ by activating LATS1/2 and by localizing YAP/TAZ to cell junctions. Conversely, loss of cell-cell contacts activates Yap/TAZ-mediated pro-tumorigenic gene expression
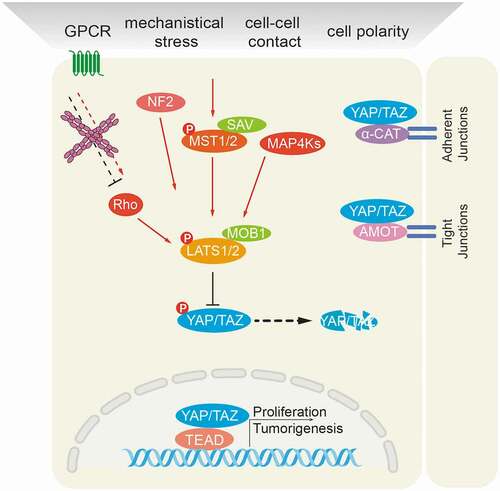
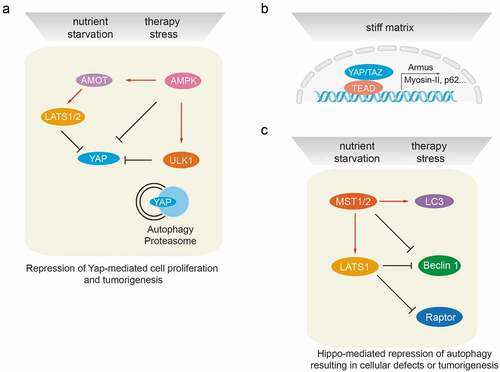
Figure 3. Cross-talk between autophagy and the Hippo signaling pathway at the molecular level. (a) Autophagy regulates Hippo signaling activity. Under starvation or therapy stress conditions, AMPK inactivates YAP by direct phosphorylation at Ser94 or by angiomotin (AMOT)/LATS1/2-mediated indirect regulation, thereby inhibiting YAP-driven oncogenic events. ULK1 is also able to phosphorylate YAP at serine residues S74/97 and to inactivate YAP. Under physiological conditions, YAP protein homeostasis is achieved by proteasomal and autophagy-mediated degradation. Hence, autophagy or proteasome deficiencies lead to an accumulation of YAP and promote YAP-driven tumorigenesis. (b) Hippo signaling mediates transcriptional regulation of autophagy. The YAP/TAZ/TEAD transcriptional complex is able to induce the expression of Armus, Myosin-II, miR-29 and p62 mRNA in stiff culture environments, thereby promoting autophagosome maturation and autophagic flux. YAP/TAZ-induced transcriptional regulation of autophagy core-components is essential to drive the cellular plasticity required for somatic stem cells as well as tumor-initiating cells. (c) Hippo signaling mediates post-transcriptional regulation of autophagy effectors. Besides activating LATS1/2, MST kinases can directly phosphorylate LC3B and promote autophagy induction, resulting in the elimination of intra-cellular bacteria. During myocardial infarction, MST1 is also able to phosphorylate BECN1 and inactivate BECN1 by inducing BECN1/BCL2 complex formation. Such inhibition of autophagy promotes cardiac dysfunction. Under cell-cell contact conditions, LATS kinases can phosphorylate Raptor at Ser606 and inhibit mTORC1 activity, leading to reduced organ size. During cancer therapy stress, in a kinase activity-independent fashion LATS1, but not LATS2, can induce K27-linked polyubiquitination of BECN1, thereby promoting BECN1 homodimer formation and inhibiting autophagic cell death. The loss of autophagy results in cellular defects and/or in tumorigenesis