Figures & data
Figure 1. TDP-43 dysregulation compromises a range of essential cellular processes through gain- and/or loss-of-function. Gain-of-function: TDP-43 cytoplasmic aggregates sequester multiple nucleoporin components of the nuclear pore complex (NPC), leading to abnormal nuclear membrane morphology and defective nucleocytoplasmic transport. Cytoplasmic TDP-43 also localizes to mitochondria, binds to mRNAs encoding respiratory chain complex I core subunits ND3 and ND6, and represses their translation. Reduced respiratory chain complex protein expression likely results in energy deficiency. TDP-43 promotes ER-mitochondria dissociation by disrupting VAPB–PTPIP51 interactions and thus decreases mitochondrial calcium uptake. Loss-of-function: Mutant TDP-43 loses interactions with XRCC4/LIG4 complex at DSB sites due to cytoplasmic mis-localization and compromises NHEJ. Notably, loss of TDP-43 leads to cryptic exons' inclusion into mRNAs that causes frameshifts and NMD. TDP-43 appears to mediate autophagosome-lysosome fusion. Loss of TDP-43 blocks the fusion process and impairs autolysosome formation
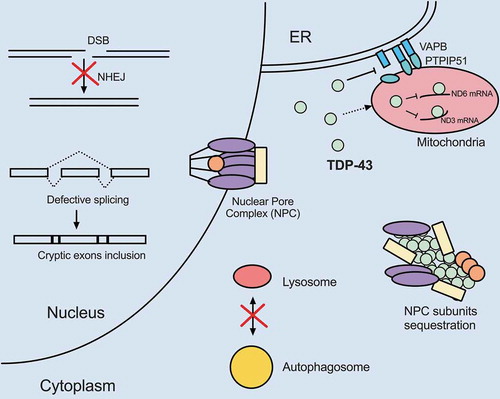
Figure 2. FUS dysregulation compromises nuclear and cytoplasmic processes shared with TDP-43. Overexpression of wild-type or expression of mutant FUS appear to disrupt pathways similar to TDP-43 dysregulation. FUS localizes to mitochondria and disrupts respiratory chain complex V assembly and results in energy deficiency. FUS gain-of-function also disrupts VAPB–PTPIP51 interactions and promotes ER-mitochondria dissociation. FUS plays physiological roles in DSB and SSB repair. Therefore, loss of nuclear FUS due to cytoplasmic mis-localization compromises DDR and increases DNA damage. FUS also participates in splicing, especially autoregulation of its own transcript level. Loss of nuclear FUS impairs autoregulation and results in increased FUS mRNA production. Mutant FUS cytoplasmic aggregates sequester spliceosome, translation and NMD factors, leading to mis-splicing of introns, pre-mature translational termination and hyper NMD. Additionally, FUS aggregates sequester numerous mRNAs including nuclear-encoded mitochondrial mRNAs and represses their translation. Deficiency in these mitochondrial components further leads to mitochondrial impairment. Finally, overexpression of wild-type or expression of mutant FUS inhibits autophagosome-lysosome fusion
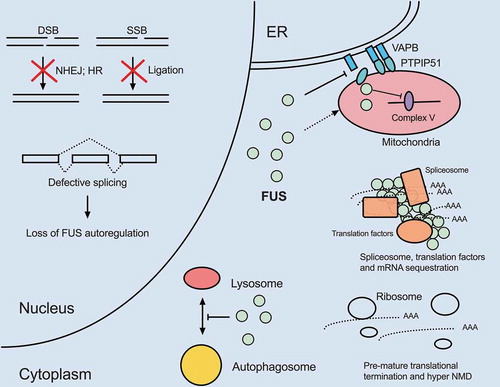
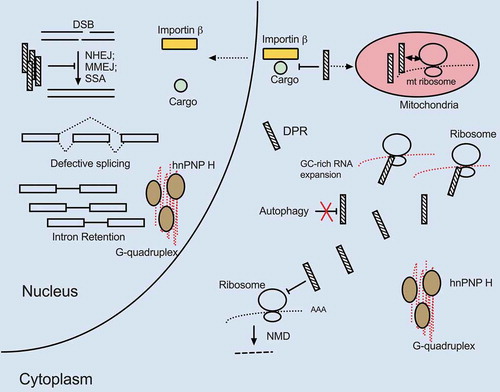
Figure 3. Mutant C9ORF72 triple hits. The GC expansion mutation within a C9ORF72 intron causes highly structured RNA G-quadruplex formation, DPR production through RAN translation and reduced C9ORF72 protein expression. All three processes are deleterious to cells and increase the likelihood of MN death. G quadruplex-containing C9 RNA sequesters hnRNP H, resulting in widespread transcripts mis-splicing, including of transcripts encoding proteasome subunits essential for protein degradation. The GC-rich intronic expansion can be translated via RAN translation, resulting in toxic DPR expression. DPRs inhibit DSB repair, NMD and prevent cargo binding of importin-ß as well as induce mitochondrial dysfunction by localizing to mitochondria and interacting with mitochondrial ribosome subunits. The GC-rich expansion in C9ORF72 inhibits transcription and can lead to overall reduction in C9ORF72 protein levels. Since C9ORF72 protein is important for autophagy, cells with lower C9ORF72 protein levels may further accumulate protein aggregates, damaged organelles and toxic DPRs