
Figures & data
Figure 1. Activation of the cGAS/STING signaling pathway. cGAS is a cytoplasmic nucleic acid sensor that recognizes double-stranded DNA from tumor cells, bacteria, and viruses in the cytoplasm. By binding to heterologous DNA to form a dimer, it induces a conformational change in itself, thereby promoting the conversion of ATP and GTP to cGAMP. As a second messenger, cGAMP binds to STING anchored in the endoplasmic reticulum, induces its conformational change, and promotes the transfer of STING from the endoplasmic reticulum to the Golgi. Subsequent recruitment of TBK1 induces IRF3 phosphorylation, which elicits type 1 IFN-mediated immune responses. In addition, STING can also activate IκB kinase, phosphorylate IκB, release NF-κB into the nucleus, and induce the expression of IFN and inflammatory cytokines. ①: Ribonucleoprotein A2B1 promotes the nucleocytoplasmic transport of cGAS, IFI16, and STING through N6-methyladenosine modification, thereby forming a positive feedback loop for TANK/ TBK1/ IRF3 activation. ② the Yip family and STING ER exit protein recruit STING to COPII. STEEP promotes VPS34 complex-mediated phosphatidylinositol-3-phosphate production and ER membrane bending by binding to STING and recruits the curvature-binding protein Sar1 to promote COPII-mediated ER transport of STING to the Golgi apparatus. ③: TBK1 enhances the affinity of p62 for ubiquitinated STING through the phosphorylation of p62 and promotes its transport to autophagosomes for degradation. ④: the interferon gamma-inducible protein 16 acts on STING through the pyrin domain and cooperates with cGAMP to promote the phosphorylation and translocation of STING.
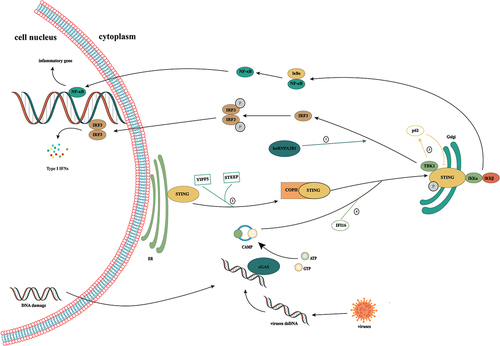
Figure 2. Activation and inhibition of cGAS/STING signaling in tumor immunity. In tumor immunity, dsDNA generated by gene replication of tumor cells can activate the cGAS/STING pathway, in which dsDNA can be delivered to APC cells through extracellular vesicles or gap junctions, and GAMP can be delivered to APC cells through cGAMP exporters. APC activates its own cGAS/STING by recognizing this dsDNA and cGAMP to induce a type I IFN-mediated immune response, thereby recruiting CD8+ cells and NK cells to mediate tumor cell apoptosis. ①: STING expression is epigenetically inhibited by histone H3K4 lysine demethylase KDM5B and KDM5C, which inhibit cGAS/STING signaling by maintaining low H3K4me3 levels in the STING promoter region. ②: high expression of DNMT1 and EZH2 was found to lead to significant silencing of STING expression and inhibition of cGAS/STING signaling. ③: receptor tyrosine kinase HER2 binds to the C-terminal domain of STING through its domain, recruits AKT1 to STING and phosphorylates TBK1S510 through AKT1 to prevent the TBK1/STING and TBK1K63-ubiquitin associations. ④: Tumor cells inhibit STING activation by upregulating exonuclease ENPP1 expression and hydrolyzing the immune transmitter cGAMP. ⑤: Tumor cells promote STING degradation in a SOX2 autophagy-dependent manner.
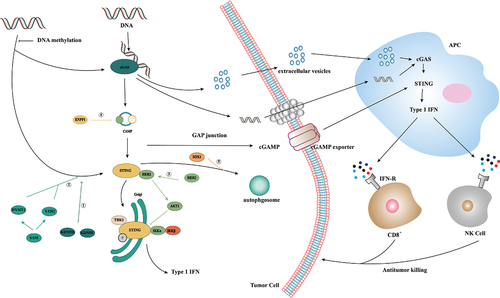
Figure 3. cGAS/STING signal activation promotes tumor immunity. Since cGAS-STING signaling is generally inhibited in tumor cells, activation of the cGAS-STING signaling pathway through multiple pathways is an important idea for tumor therapy. ① cdiGMP, c-diAmp,cgamp, and 2′, 3′-cGAMP are human natural cyclic dinucleotides (CDN), which can effectively induce inflammation and tumor necrosis by intratumoral injection. ② DSDP and BNBC are small-molecule STING agonists that can induce the activation of the cGAS/STING signaling pathway. ③ RR-S2 CDA and MLRR-S2 CDA are CDN derivatives, which have the same effect as natural CDA, but their stability in vivo is better. ④ It activates the cGAS-STING signaling pathway by increasing the DNA damage of tumor cells and inducing abnormal DNA aggregation in the cytoplasm. For example, telomere targeting drugs 6-thio-20-deoxyguanosine and doxorubicin are used.
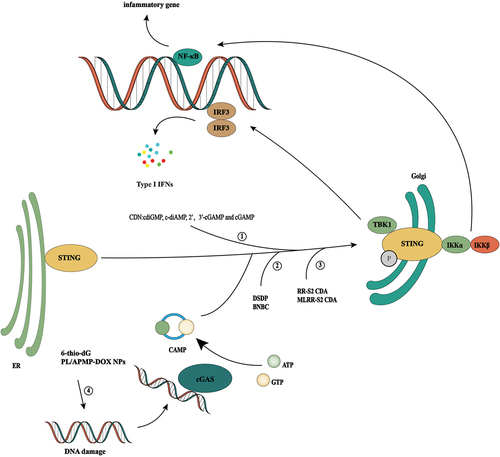
Figure 4. Activation and inhibition of cGAS/STING signaling in viral infection. During viral infection, cells recognize viral DNA through cGAS and activate the cGas/sting pathway, thereby exerting antiviral immunity. However, viruses have evolved a variety of mechanisms to evade host immunity in the confrontation with humans. ① hbx interferes with cGas’s recognition of DNA by interacting with cGAS. But HBx was found to upregulate TBK1 expression at the transcriptional level, thus promoting the occurrence of HCC by activating NF-κB. ② HPV16 E7 can mediate transcriptional activation of chromatin repressor SUV39H1, induce the formation of heterochromatin in the promoter region of the STING gene and effectively inhibit the intracellular cGAS/STING signaling pathway. ③: BFRF1 can evade the innate immunity of the host by inhibiting IRF3 activation by interacting with the downstream signal IKKi of the cGAS/STING pathway. ④: in KSHV infection, the cytoplasmic LANA variant inhibits the production of cGAS/STING-dependent interferon through direct interaction with cGAS. Meanwhile, Kaposi’s sarcoma virus ORF52 directly inhibits the activity of the cGAS enzyme by binding to cGAS and DNA. ⑤: vIRF1 targets STING by blocking the interaction between STING and TBK1, thus inhibiting the phosphorylation and concomitant activation of STING and ultimately inhibiting the DNA induction pathway. ⑥: by binding to STING, CMV protein M152 slows its transport to the Golgi chamber and inhibits STING from initiating type I IFN signaling in the Golgi chamber. ⑦: HTLV-1 interacts with STING through the protein Tax and reduces ubiquitination of its K63 connection, resulting in a decrease in the STING/TBK1 association and IFN-I production. Meanwhile, Tax-1 directly targets the autophagy complex containing Beclin1, which relieves the regulation of autophagy by stimulating the IKK complex, thus leading to continuous activation of NF-kB.
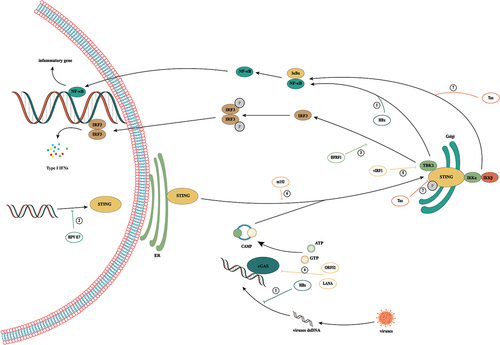
Figure 5. cGAS/STING signaling pathway and cellular senescence. CCFs can be induced by dsDNA generated in senescent cells due to oncogene activation, telomere dysfunction, and chromosomal instability. cGAS activates the cGAS/STING pathway by recognizing CCFs and triggers the production of SASP factors, thereby promoting the recruitment of immune cells to senescent cells, and inducing the clearance of senescent cells through immune cell action. ①: ROS drives the formation of CCFs through ROS/JNK retrograde signaling pathways. ②: cGAS, as an autophagy receptor, binds to LC3B through its LC3 interaction region and promotes the transfer of autophagy micronuclei to lysosomes in an ATG14 and ATG7 dependent manner, which inhibits micronucleus-driven cGAS activation and subsequent cGAMP production. ③: TLR2 promotes cell cycle arrest by regulating the tumor inhibitor and regulates the SASP by inducing serum A-SAA in the acute phase. ④: STING can regulate the senescence microenvironment through DOT1L-mediated H3K79 methylation at the IL1A site in OIS cells.
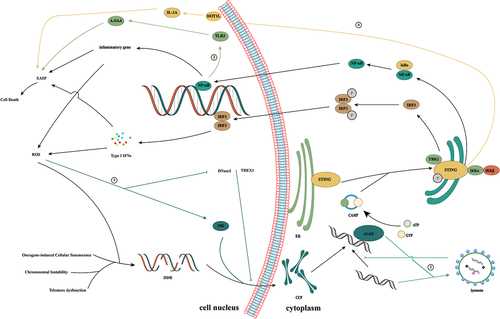
Figure 6. The cGAS/STING signaling pathway promotes tumor progression. Tumor cells are often filled with cytosolic dsDNA. Normally, tumor growth can be inhibited by inducing the cGAS/STING signaling pathway. However, studies have shown that in some cases, activation of the cGAS/STING signaling pathway can induce the generation of an inflammatory tumor microenvironment, thereby inducing tumorigenesis. ① STING can promote tumor growth by inducing IDO responses in the tumor microenvironment. ② DMBA activates STING-dependent signaling with intrinsic inflammatory effects that induce tumorigenesis. ③ Tumor cells selectively establish gap junctions with astrocytes through PCDH7, transfer tumor cells to cGAMP to astrocytes, activate the STING pathway, and produce inflammatory cytokines IFNα and TNFα. These inflammatory factors secreted by astrocytes can activate STAT1 and NF-κB pathways in tumor cells, thereby supporting tumor growth and chemotherapy resistance. ④ CIN can activate the cGAS/STING signaling pathway in tumor cells, thereby activating the inflammation-related tumor microenvironment to promote tumor progression.
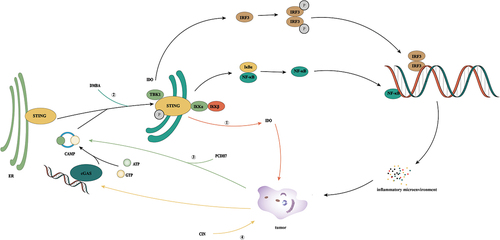
Figure 7. The crosstalk of cGAS/STING pathway in infection, senescence, and tumor. ① In the face of tumor formation, cells induce tumor cell senescence through the cGAS/STING signaling pathway. ② Under specific circumstances, the cGAS/STING signaling pathway can induce the generation of an inflammatory microenvironment, which can lead to the occurrence of inflammation-related tumors. ③ During virus infection, on the hand, it inhibits the cGAS/STING activity of host cells by inhibiting the activities of cGAS, STING, and IRF3, on the other hand, it promotes the release of inflammatory factors by activating NF-κB, and at the same time induces the production of an inflammatory microenvironment through self-reproduction, which induces tumorigenesis.
Data availability statement
Data sharing does not apply to this article as no new data were created or analyzed in this study.