Figures & data
Figure 1. RNAomeSeq set up and analysis. (A). Diagram of biological replicate sample preparation from mES cells treated with 2.7 μM cisplatin or mock-treated (equal volume DMSO) for 8 hours. This procedure was repeated 4 times to obtain 4 independent biological replicates. All omics methods were performed on the exact same samples. (B). Schematic of the RNAomeSeq method. Total RNA was depleted of rRNA, fragmented and adapters were ligated to prepare a compatible cDNA library followed by fractionation on gel. Short sequencing reads (<36 nucleotides) were trimmed for adapter sequences and further processed by TRAP (). 36 nucleotide sequencing reads were processed as long RNAs.
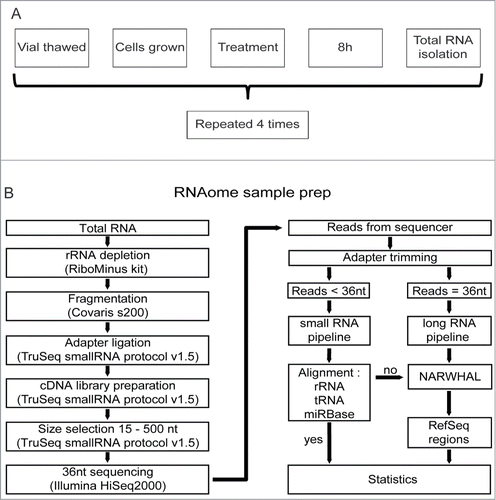
Figure 2. Schematic of the Total RNA Analysis Pipeline, TRAP, for analysis of sequencing datasets. (A). Modules for long RNA analysis, script 1 for RefSeq annotated exonic transcripts and script 2 for RefSeq annotated non-exonic regions. (B). Modules for small RNA analysis, script 3 to align trimmed reads to first rRNA, then tRNA sequences and the microRNA database, miRBase version 19.
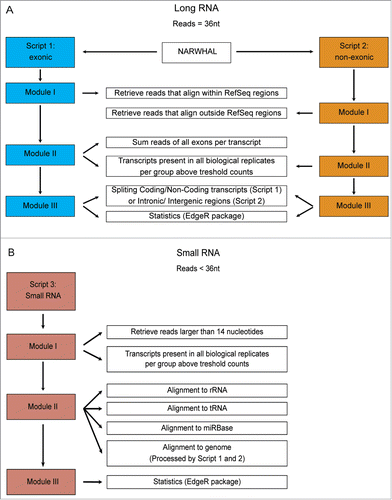
Figure 3 (See previous page). The proportion of RNA species found in mES cells. (A). The proportion of RNA classes detected by the RNAomeSeq protocol with a minimum of one read per million found across all biological replicates from at least one of the experimental groups. Detecting small RNA classes (right panel): tRNA fragments (0.2%), small coding (0.2%), small non-coding (0.3%), mature microRNA (miR) (0.7%), microRNA isoforms (isomiR) (0.9%), small intergenic (1.7%), small intronic (2.0%); and long RNA classes (left panel): non-coding transcripts also containing complete tRNAs (12.2%), coding transcripts (2.2%), snoRNA (19.4%), mitochondrial (1.9%), histones (0.2%), intronic region (37.4%), intergenic region (20.7%) classes. (B). The proportion of RNA species detected by the mRNASeq protocol with a minimum of 5 reads found across all biological replicates from at least one of the experimental groups. Detecting coding transcripts (71.0%), non-coding transcripts (1.2%) and reads from mitochondrial (2.3%), histones (0.1%), intronic regions (9.3%) and intergenic regions (16.2%). (C). The proportion of small RNA species detected by the smallRNASeq protocol with a minimum of 5 reads found across all biological replicates from at least one of the experimental groups. Detecting small RNA classes: tRNA fragments (4.0%), small coding (2.0%), small non-coding (17.6%), mature microRNA (miR) (27.9%), microRNA isoforms (isomiR) (25.7%), small intergenic (10.6%) and small intronic (12.1%). The indicated % represents the total aligned RNAs from that particular class compared to the total number of reads, excluding rRNA reads.
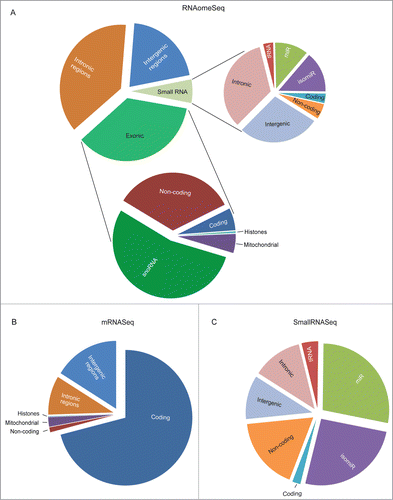
Figure 4. Representation of coding transcripts. (A) Coding transcript length distribution of the whole genome or detected by mRNASeq and RNAomeSeq. (B) The Pearson-correlation between and X-Y scatter plot of coding transcript expression between RNAomeSeq and mRNASeq, histones encircled in red. (C) Distribution of reads along the body of all coding transcript for mRNASeq, RNAomeSeq and RNAomeSeq depl (depleted of histones and transcripts with intronic snoRNA). (D) Distribution of reads aligning to the detected coding transcripts by mRNASeq, RNAomeSeq and RNAomeSeq depl (depleted of histones and transcripts with intronic snoRNA) in regard to transcript length.
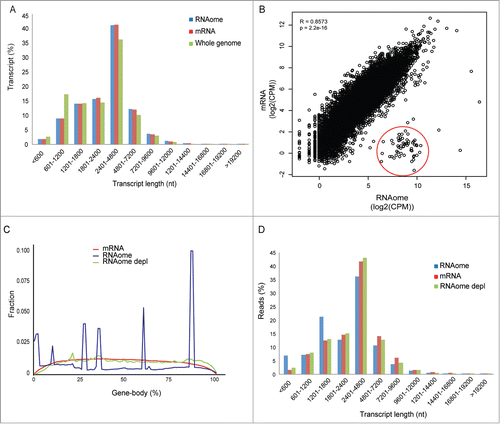
Figure 5. Representation of microRNAs and isomiRs. (A) Length distribution of the microRNA/isomiRs transcripts in the miRbase database or detected by smallRNASeq and RNAomeSeq. (B) The Pearson-correlation between and X-Y scatter plot of microRNA/isomiRs expression between RNAomeSeq and smallRNASeq. (C) Distribution of microRNA/isomiRs reads detected by smallRNASeq and RNAomeSeq in regard to length.
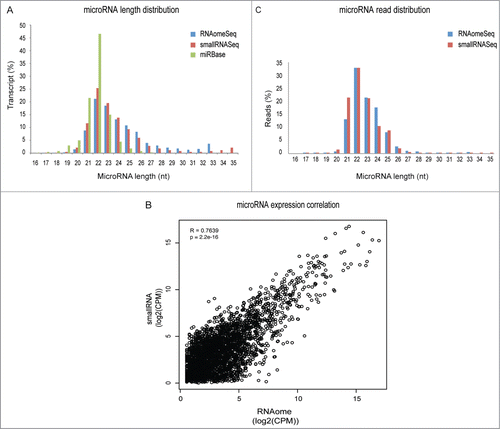
Table 1. The Pearson-correlation between replicate samples in RNAomeSeq, mRNASeq and smallRNASeq, for the coding transcripts and/or microRNAs, all correlations had P-value < 2.2E–16.
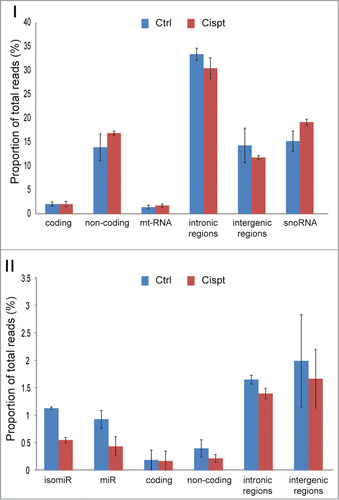
Figure 6. Quantitative preservation of all RNA species. Total proportion of RNA classes before and after cisplatin treatment. Panel I, long RNA classes, Panel II small RNA classes. Error bars represent standard deviations.