
Figures & data
Figure 1. Ribozyme-processed crRNA enhances the Luc knockdown activity of CRISPR-Cpf1. (a) The crRNA structures of the As and LbCpf1 systems. Both crRNA molecules consist of a ~ 20-nt scaffold and a 23-nt guide (N23). The variable U1-6 tail at the 3ʹ-end is generated when a standard Pol III promoter cassette is used. The variable loop nucleotide positions are marked in blue and green boxes. The first crRNA nucleotide is marked as +1A, but the upstream U (in brackets) has also been implicated in the transcription initiation process. (b) Schematic of three crRNA expression constructs. The Pol III human U6 promoter drives crRNA transcription up to the T6 (TTTTTT) termination signal. The HH and HDV ribozymes were introduced to guide crRNA processing exacty at the crRNA border (marked as scissor). The +1A represents the first crRNA nucleotide. (c) Luc knockdown activity of CRISPR-Cpf1. An equimolar amount of crLuc constructs (equivalent to 50 ng cr vector) together with their cognate Cpf1 plasmids (equivalent to 100 ng AsCpf1 vector) were co-transfected into HEK293T cells with 200 ng Luc reporter and 2 ng Renilla luciferase plasmid to control for the transfection efficiency. The empty cr plasmid served as negative control. The relative Luc activity normalized for Renilla expression was determined at two days post-transfection and the control Luc activity was arbitrarily set at 100%. The results are shown as mean values ± standard deviation (SD, n = 3).
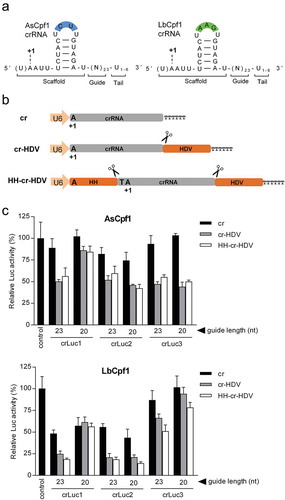
Figure 2. Titration of CRISPR-Cpf1 components. CRISPR-Cpf1 constructs were titrated in the 37.5–600 ng range and co-transfected into HEK293T cells with 200 ng Luc reporter and 2 ng Renilla luciferase plasmid. The titration was performed with a fixed 1:2 stoichiometry of the cr plasmid and the matching Cpf1 plasmid. The relative Luc activity was determined two days post-transfection and all data are presented as the mean value ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; one-way ANOVA.
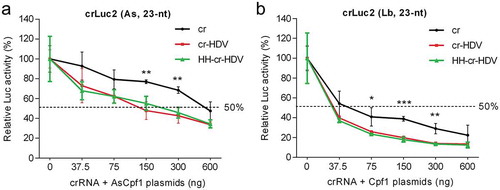
Figure 3. crRNA expression from the crRNA-ribozyme cassettes. HEK293T cells in a 6-well plate were transfected with the same molar amount of indicated crRNA constructs (equivalent to 1 µg cr vector) and a fixed molar amount of their cognate Cpf1 plasmids (equivalent to 2 µg AsCpf1 vector). Total cellular RNA was harvested two days post-transfection and 5 µg was subjected to Northern blotting using the Luc2 probe targeting the crLuc2 guide region (panel A) and the Lb probe targeting Lb crRNA scaffold (panel B). The crRNA and precursor crRNA-HDV transcripts are marked. An RNA size marker (nt) was included. Ethidium bromide staining of 5S RNA is shown at the bottom as loading control. Quantitation of crRNA normalized by 5S RNA is plotted below the blot and the cr signal of crLuc2 (As) was arbitrarily set at 10. The results were produced in two independent experiments that showed similar trends.
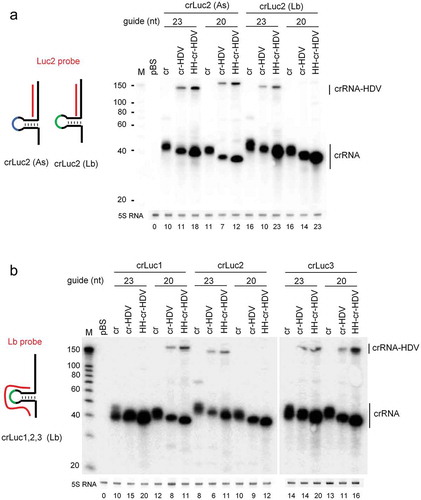
Figure 4. The cr-HDV design enhances the gene editing efficiency. (a) Comparison of the cr and cr-HDV constructs for chromosomal Luc gene editing. The same molar amounts of cr and cr-HDV plasmids (equivalent to 100 ng cr vector) and 200 ng LbCpf1 plasmid were co-transfected into HeLa X1/6 cells. Two days post-transfection, the cells were harvested for the Surveyor nuclease assay to estimate the Luc editing efficiency. The empty cr construct served as negative control. The unedited Luc gene amplicon is marked by an open triangle and the black triangles mark the Surveyor nuclease-cleaved bands due to Luc gene editing by Cpf1. The indel percentage (%) was calculated and is plotted below. The graph represents one of two independent experiments that yielded similar results. (b) Comparison of cr and cr-HDV for CCR5 gene editing. An equimolar amount of cr/cr-HDV plasmid (equivalent to 100 ng cr) and 200 ng LbCpf1 plasmid were co-transfected into TZM-bl cells with 100 ng JS-1 plasmid (a GFP expression vector) serving as transfection control. Two days post-transfection, cells were stained with anti-CCR5 antibody and the CCR5 expression was determined by FACS analysis. The empty cr construct servesd as negative control. The y axis represents forward-scattered light (SSC) and the x axis represents CCR5 fluorescence intensity. The CCR5 disruption efficiency was measured for the GFP-positive cell fraction. The fold increase (×) in editing efficiency of cr-HDV versus cr was calculated. The results are presented as mean value ± SD of three independent experiments. The raw data are shown in Figure S1.
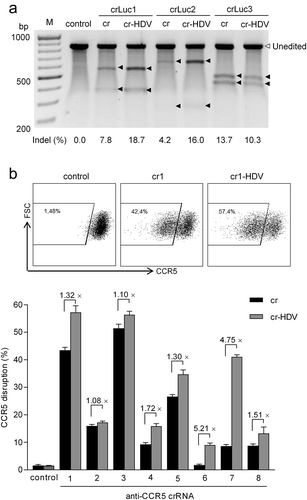
Figure 5. The cr-HDV design also increases the CRISPRa activity. (a) Schematic of the dCpf1-VP64 construct. (b) Schematic of Tet-On inducible Luc reporter cassette (7xTRE-CMVminimal-Luc) that is chromosomally integrated in HeLa X1/6 cells. The TRE and the location of the crRNA targets are indicated, crRNA1-4 target the 7× TRE repeat and crRNA5 and 6 the Luc gene leader sequence. (c) Luc induction by the CRISPR-based systems. Dox addition and transfection of a rtTA expressing plasmid was used as positive control. For CRISPR-based Tet-On induction, an equimolar amount of cr/cr-HDV construct (equivalent to 100 ng cr vector) and a fixed amount of the cognate dCpf1-VP64 plasmid (equivalent to 200 ng dAsCpf1-VP64 vector) were transfected into HeLa X1/6 cells with a Renilla plasmid to control for transfection efficiency. The empty crRNA plasmid acted as negative control. The relative Luc expression normalized by Renilla was determined at two days post-transfection. The results are represented as mean value ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; unpaired t test.
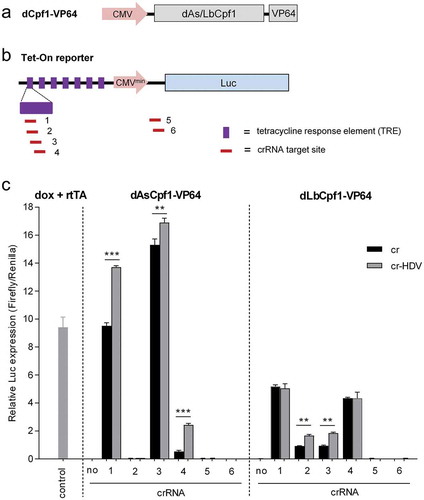
Figure 6. The least active crRNAs benefit the most from the HDV design. We plotted the fold increase by the HDV insertion versus the relative crRNA activity (%) of the original cr construct. The data on gene editing and gene activation was taken from and , respectively. The Pearson’s correlation coefficient (r) was calculated.
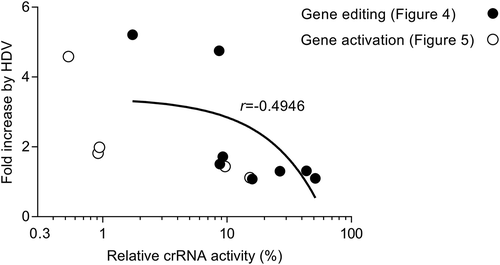
Table 1. Target Sequence (PAM Underlined).