
Figures & data
Figure 1. Depletion of the nucleolar RNA helicase DHX37 affects early and late stages of 18S rRNA maturation. (a) HeLa cells were fixed and the localization of DHX37 was determined by immunofluorescence using an antibody against endogenous DHX37 (green). Immunofluorescence against NSUN5 (red) served as a nucleolar marker and nuclear material was visualized by DAPI staining (blue). An overlay of the images is provided and a scale bar representing 10 μm is depicted. (b) HeLa cells were left untransfected (WT), or were transfected with non-target siRNA (siNT) or siRNAs targeting DHX37 (siDHX37_1 and siDHX37_2). After 72 h, cells were harvested, and proteins were analyzed by western blotting using antibodies against DHX37 and tubulin. (c) Whole cell extracts from cells treated with non-target siRNAs (siNT) or a siRNA targeting DHX37 (siDHX37_1) were separated by sucrose density gradient centrifugation. The absorbance of each fraction at 260 nm was used to generated a profile on which the positions of the ribosomal and pre-ribosomal complexes are indicated. (d) Schematic view of the major pre-rRNA intermediates detected in human cells. Mature rRNA regions are shown as black rectangles, and internal (ITS) and external transcribed spacer (ETS) regions are represented by black lines. Cleavage sites relevant for maturation of the 18S rRNA are named above the 47S rRNA transcript and the hybridization position of the 5ʹ ITS1 probe used for northern blotting is indicated by the triangle. (e) Untransfected HeLa cells, or HeLa cells that had been treated with non-target siRNAs (siNT) or those targeting DHX37 (siDHX37_1) for 72 h were starved of phosphate then grown in media supplemented with [32P]-orthophosphate for 1 h before growth in normal DMEM for 3 h. Total RNA was extracted, separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and nascent RNAs were detected using a phosphorimager. The mature 28S rRNA was visualized using methylene blue staining (MB). (f) Total RNA was extracted from wild-type (WT) HeLa cells or HeLa cells transfected with siRNAs as in (b). RNAs were separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was visualized by methylene blue staining (MB). Northern blotting with a probe hybridizing to the 5ʹ end of ITS1 (5ʹ ITS1) was used to detect precursors of the 18S RNA, which were visualized using a phosphorimager. Aberrant pre-rRNA species detected upon depletion of DHX37 are indicated by arrows. Experiments were performed at least in triplicate and representative data are shown.
![Figure 1. Depletion of the nucleolar RNA helicase DHX37 affects early and late stages of 18S rRNA maturation. (a) HeLa cells were fixed and the localization of DHX37 was determined by immunofluorescence using an antibody against endogenous DHX37 (green). Immunofluorescence against NSUN5 (red) served as a nucleolar marker and nuclear material was visualized by DAPI staining (blue). An overlay of the images is provided and a scale bar representing 10 μm is depicted. (b) HeLa cells were left untransfected (WT), or were transfected with non-target siRNA (siNT) or siRNAs targeting DHX37 (siDHX37_1 and siDHX37_2). After 72 h, cells were harvested, and proteins were analyzed by western blotting using antibodies against DHX37 and tubulin. (c) Whole cell extracts from cells treated with non-target siRNAs (siNT) or a siRNA targeting DHX37 (siDHX37_1) were separated by sucrose density gradient centrifugation. The absorbance of each fraction at 260 nm was used to generated a profile on which the positions of the ribosomal and pre-ribosomal complexes are indicated. (d) Schematic view of the major pre-rRNA intermediates detected in human cells. Mature rRNA regions are shown as black rectangles, and internal (ITS) and external transcribed spacer (ETS) regions are represented by black lines. Cleavage sites relevant for maturation of the 18S rRNA are named above the 47S rRNA transcript and the hybridization position of the 5ʹ ITS1 probe used for northern blotting is indicated by the triangle. (e) Untransfected HeLa cells, or HeLa cells that had been treated with non-target siRNAs (siNT) or those targeting DHX37 (siDHX37_1) for 72 h were starved of phosphate then grown in media supplemented with [32P]-orthophosphate for 1 h before growth in normal DMEM for 3 h. Total RNA was extracted, separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and nascent RNAs were detected using a phosphorimager. The mature 28S rRNA was visualized using methylene blue staining (MB). (f) Total RNA was extracted from wild-type (WT) HeLa cells or HeLa cells transfected with siRNAs as in (b). RNAs were separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was visualized by methylene blue staining (MB). Northern blotting with a probe hybridizing to the 5ʹ end of ITS1 (5ʹ ITS1) was used to detect precursors of the 18S RNA, which were visualized using a phosphorimager. Aberrant pre-rRNA species detected upon depletion of DHX37 are indicated by arrows. Experiments were performed at least in triplicate and representative data are shown.](/cms/asset/a169d729-831c-41ba-87ed-2aa8ef4fc227/krnb_a_1556149_f0001_c.jpg)
Figure 2. Mapping of the aberrant 16S* and 38S* pre-rRNA intermediates that accumulate upon depletion of DHX37. (a) Schematic view of the 47S pre-rRNAs (black) and the aberrant 38S*, 16S* and 30SL5ʹ pre-rRNA species not normally detected in human cells (grey). Mature rRNA regions are shown as rectangles, and internal (ITS) and external transcribed spacer (ETS) regions are represented by lines. Cleavage sites are named above the 47S pre-rRNA and the hybridization position of probes used for northern blotting are indicated. (b,c) RNA extracted from HeLa cells transfected with non-target siRNAs (siNT) or siRNAs targeting DHX37 (siDHX37_1) was separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was detected by methylene blue staining (MB). Northern blotting was performed using probes hybridizing to different positions within the 5ʹ ETS and ITS1 (b) or the 18S rRNA (c) and pre-rRNAs were visualized using a phosphorimager. (d) RNA and proteins were extracted from wildtype (WT) HeLa cells or cells that had been transfected with non-target siRNAs (siNT), or siRNAs targeting DHX37 (siDHX37_1) or XRN2 (siXRN2). Proteins were analyzed by western blotting using the antibodies indicated to the right (upper panel) and pre-RNAs were detected by northern blotting using a probe hybridizing to the 5' end of ITS1.
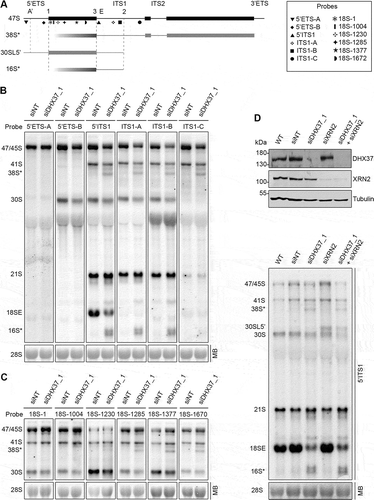
Figure 3. Pre-rRNAs are degraded upon depletion of DHX37 and expression of catalytically inactive DHX37 leads to defects in A’ cleavage and the conversion of 21S to 18SE. (a) C-terminally His6-tagged DHX37 or DHX37 carrying a threonine to alanine substitution at amino acid 282 within the evolutionarily conserved ‘GKT’ motif (DHX37T282A) was recombinantly expressed in E. coli and purified. Purified proteins were separated by SDS-PAGE and visualized by Coomassie staining. (b) The amount of ATP hydrolyzed by recombinant DHX37 or DHX37T282A in the presence (+) or absence (-) of RNA was determined using an in vitro NADH-coupled ATPase assay. Experiments were performed in triplicate and error bars represent mean ± standard deviation. (c) HEK293 cell lines were transfected with either non-target siRNAs (siNT) or siRNAs targeting DHX37 (siDHX37_1) and expression of the Flag tag, or C-terminally Flag-tagged DHX37 or DHX37T282A was induced by addition of tetracycline 24 h before harvesting. Proteins were extracted and analyzed by western blotting using antibodies against DHX37, tubulin and the Flag tag. (d) Total RNA extracted form HEK293 cell lines treated as described in (c) was separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and analyzed by northern blotting using probes hybridizing at the 5ʹ end of ITS1 (left panel) or within the 5ʹ ETS (right panel). Pre-rRNAs were detected using a phosphorimager and the mature 28S rRNA was visualized by methylene blue staining (MB).
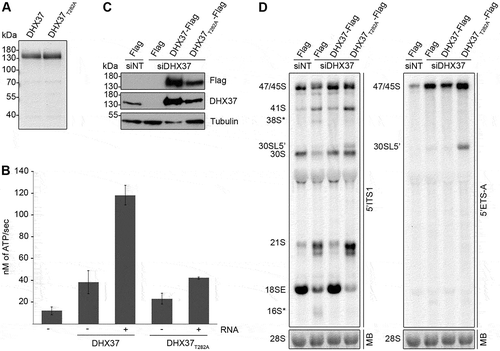
Figure 4. DHX37 associates with the 3ʹ region of the U3 snoRNA that contains the box C/D motifs. (a) HEK293 cells expressing DHX37-Flag or the Flag tag were grown in the presence of 4-thiouridine before crosslinking in vivo using light at 365 nm. RNA-protein complexes were tandem affinity purified under native and denaturing conditions, then RNAs co-purified with DHX37 were ligated to sequencing adaptors and labelled using [32P]. Complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and labelled RNAs were detected by autoradiography. The regions of the membrane excised for subsequent analysis are indicated by boxes. (b) The region of the membrane containing DHX37-Flag-RNA complexes as indicated in (a), and a corresponding region of the lane containing the Flag sample, were excised. RNAs were isolated and subjected to reverse transcription and PCR amplification to generate a cDNA library upon which Illumina deep sequencing was performed. The obtained sequence reads were mapped to the human genome and, after normalization, the relative number of reads derived from snoRNAs in the Flag and DHX37-Flag samples was determined. (c) The relative distribution of sequence reads derived from each box C/D snoRNA in the Flag and DHX37-Flag datasets is shown. The proportion of sequence reads mapping to the U3 snoRNA is highlighted in red while all other snoRNAs are shown in shades of grey. (d) Cell extracts prepared from HEK293 cells expressing Flag-DHX37 or the Flag tag were incubated with anti-Flag beads. After thorough washing steps, complexes were eluted and RNA was extracted. RNAs extracted from inputs (1%) and eluates were separated by denaturing PAGE, transferred to a nylon membrane and northern blotting was performed using probes hybridizing to the U3 and U8 snoRNAs. RNAs were detected using a phosphorimager. (e) The normalized number of reads mapping to each nucleotide of the gene encoding the U3 snoRNA in the Flag (black) and DHX37-Flag (red) datasets is shown graphically (upper panel). The normalized number of mutations mapping to each position are indicated in the lower panel. A schematic representation of the U3 snoRNA is shown below with the relative positions of key features indicated.
![Figure 4. DHX37 associates with the 3ʹ region of the U3 snoRNA that contains the box C/D motifs. (a) HEK293 cells expressing DHX37-Flag or the Flag tag were grown in the presence of 4-thiouridine before crosslinking in vivo using light at 365 nm. RNA-protein complexes were tandem affinity purified under native and denaturing conditions, then RNAs co-purified with DHX37 were ligated to sequencing adaptors and labelled using [32P]. Complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and labelled RNAs were detected by autoradiography. The regions of the membrane excised for subsequent analysis are indicated by boxes. (b) The region of the membrane containing DHX37-Flag-RNA complexes as indicated in (a), and a corresponding region of the lane containing the Flag sample, were excised. RNAs were isolated and subjected to reverse transcription and PCR amplification to generate a cDNA library upon which Illumina deep sequencing was performed. The obtained sequence reads were mapped to the human genome and, after normalization, the relative number of reads derived from snoRNAs in the Flag and DHX37-Flag samples was determined. (c) The relative distribution of sequence reads derived from each box C/D snoRNA in the Flag and DHX37-Flag datasets is shown. The proportion of sequence reads mapping to the U3 snoRNA is highlighted in red while all other snoRNAs are shown in shades of grey. (d) Cell extracts prepared from HEK293 cells expressing Flag-DHX37 or the Flag tag were incubated with anti-Flag beads. After thorough washing steps, complexes were eluted and RNA was extracted. RNAs extracted from inputs (1%) and eluates were separated by denaturing PAGE, transferred to a nylon membrane and northern blotting was performed using probes hybridizing to the U3 and U8 snoRNAs. RNAs were detected using a phosphorimager. (e) The normalized number of reads mapping to each nucleotide of the gene encoding the U3 snoRNA in the Flag (black) and DHX37-Flag (red) datasets is shown graphically (upper panel). The normalized number of mutations mapping to each position are indicated in the lower panel. A schematic representation of the U3 snoRNA is shown below with the relative positions of key features indicated.](/cms/asset/e067fa95-388a-4972-9dbe-0262686a9963/krnb_a_1556149_f0004_c.jpg)
Figure 5. The U3 snoRNA accumulates on pre-ribosomes upon expression of catalytically inactive DHX37. (a) HEK293 cells capable of expression of DHX37-Flag or DHX37T282A-Flag were transfected with siRNAs against DHX37 and 24 h prior to harvesting, expression of the tagged proteins was induced by addition of tetracycline. To confirm equal expression levels, proteins were analyzed by western blotting using antibodies against DHX37 and tubulin. (b) Whole cell extracts prepared from HEK293 cells treated as in (a) were separated by sucrose density gradient centrifugation. The optical density of each faction at 260 nm was determined and used to generate a profile on which the peaks corresponding to ribosomal and pre-ribosomal complexes are indicated. RNA extracted from the gradient fractions described in (b) was separated by denaturing PAGE and transferred to a nylon membrane. Northern blotting was performed using a [32P]-labelled probe hybridizing to the U3 snoRNA. The experiments shown in this figure were performed in triplicate and representative data are shown.
![Figure 5. The U3 snoRNA accumulates on pre-ribosomes upon expression of catalytically inactive DHX37. (a) HEK293 cells capable of expression of DHX37-Flag or DHX37T282A-Flag were transfected with siRNAs against DHX37 and 24 h prior to harvesting, expression of the tagged proteins was induced by addition of tetracycline. To confirm equal expression levels, proteins were analyzed by western blotting using antibodies against DHX37 and tubulin. (b) Whole cell extracts prepared from HEK293 cells treated as in (a) were separated by sucrose density gradient centrifugation. The optical density of each faction at 260 nm was determined and used to generate a profile on which the peaks corresponding to ribosomal and pre-ribosomal complexes are indicated. RNA extracted from the gradient fractions described in (b) was separated by denaturing PAGE and transferred to a nylon membrane. Northern blotting was performed using a [32P]-labelled probe hybridizing to the U3 snoRNA. The experiments shown in this figure were performed in triplicate and representative data are shown.](/cms/asset/0a0e4784-396b-4c5a-b3d8-d197f65735fa/krnb_a_1556149_f0005_b.gif)
Figure 6. UTP14A interacts with DHX37 and stimulates its ATPase activity in vitro. (a) Cell extracts prepared from HEK293 cells expressing either DHX37-Flag or the Flag tag were used for immunoprecipitation experiments in the presence (+) or absence (-) of RNase. Inputs (1%) and eluates were separated by SDS-PAGE and analyzed by western blotting using antibodies against DHX37, UTP14A, WBSCR22 and tubulin. (b) Recombinant DHX37-His6 was incubated with either IgG sepharose (beads) or with IgG sepharose on which ZZ-UTP14A-His10 had been immobilized. After washing steps, proteins were eluted, and inputs and eluates were separated by SDS-PAGE then analyzed by western blotting using antibodies against DHX37 and UTP14A. (c) The amount of ATP hydrolyzed by recombinant DHX37 in the presence (+) or absence (-) of RNA and the presence (+) or absence (-) of UTP14A was determined using an in vitro NADH-coupled ATPase assay. Experiments were performed in triplicate and error bars represent mean ± standard deviation. (d) HeLa cells were left untransfected (WT), or were transfected with non-target siRNA (siNT) or siRNAs targeting UTP14A (siUTP14A_1 and siUTP14A_2). After 72 h, cells were harvested, and proteins were analyzed by western blotting using antibodies against UTP14A (upper panel) and tubulin (lower panel). (e) Total RNA was extracted from wild-type (WT) HeLa cells or HeLa cells transfected with siRNAs as in (D). RNAs were separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was visualized by methylene blue staining (MB). Northern blotting with a probe hybridizing to the 5ʹ end of ITS1 (5ʹ ITS1) was used to detect precursors of the 18S RNA, which were visualized using a phosphorimager. (f) Whole cell extracts prepared from HEK293 cells treated with control siRNAs (siNT) or siRNAs targeting UTP14A (siUTP14A_1) were separated by sucrose density gradient centrifugation. The absorbance of each faction at 260 nm was determined and used to generate a profile on which the peaks corresponding to ribosomal subunits (40S and 60S) and 80S monosomes are indicated. RNA extracted from the gradient fractions was separated by denaturing PAGE, transferred to a nylon membrane and northern blotting was performed using a [32P]-labelled probe hybridizing to the U3 snoRNA. Replica experiments have been performed and representative data is shown in this figure.
![Figure 6. UTP14A interacts with DHX37 and stimulates its ATPase activity in vitro. (a) Cell extracts prepared from HEK293 cells expressing either DHX37-Flag or the Flag tag were used for immunoprecipitation experiments in the presence (+) or absence (-) of RNase. Inputs (1%) and eluates were separated by SDS-PAGE and analyzed by western blotting using antibodies against DHX37, UTP14A, WBSCR22 and tubulin. (b) Recombinant DHX37-His6 was incubated with either IgG sepharose (beads) or with IgG sepharose on which ZZ-UTP14A-His10 had been immobilized. After washing steps, proteins were eluted, and inputs and eluates were separated by SDS-PAGE then analyzed by western blotting using antibodies against DHX37 and UTP14A. (c) The amount of ATP hydrolyzed by recombinant DHX37 in the presence (+) or absence (-) of RNA and the presence (+) or absence (-) of UTP14A was determined using an in vitro NADH-coupled ATPase assay. Experiments were performed in triplicate and error bars represent mean ± standard deviation. (d) HeLa cells were left untransfected (WT), or were transfected with non-target siRNA (siNT) or siRNAs targeting UTP14A (siUTP14A_1 and siUTP14A_2). After 72 h, cells were harvested, and proteins were analyzed by western blotting using antibodies against UTP14A (upper panel) and tubulin (lower panel). (e) Total RNA was extracted from wild-type (WT) HeLa cells or HeLa cells transfected with siRNAs as in (D). RNAs were separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was visualized by methylene blue staining (MB). Northern blotting with a probe hybridizing to the 5ʹ end of ITS1 (5ʹ ITS1) was used to detect precursors of the 18S RNA, which were visualized using a phosphorimager. (f) Whole cell extracts prepared from HEK293 cells treated with control siRNAs (siNT) or siRNAs targeting UTP14A (siUTP14A_1) were separated by sucrose density gradient centrifugation. The absorbance of each faction at 260 nm was determined and used to generate a profile on which the peaks corresponding to ribosomal subunits (40S and 60S) and 80S monosomes are indicated. RNA extracted from the gradient fractions was separated by denaturing PAGE, transferred to a nylon membrane and northern blotting was performed using a [32P]-labelled probe hybridizing to the U3 snoRNA. Replica experiments have been performed and representative data is shown in this figure.](/cms/asset/2df503c0-945d-425c-8e94-25ebde83fd7c/krnb_a_1556149_f0006_b.gif)