
Figures & data
Figure 1. Schematic representation of the signaling in GIST. KIT and PDGFRA mutant GIST cases are characterized by gain-of-function mutations that activate the tyrosine kinase receptor in a constitutive and ligand-independent manner. KIT and PDGFRA WT GIST cases do not present gain-of-function alterations but may overexpress IGF1R. KIT, PDGFRA and IGF1R are tyrosine kinase receptors and their activation results in the promotion of downstream cascades, including PtdIns(3,4,5)P3-MTOR, JAK-JUN and RAS-MAPK/ERK, which lead to uncontrolled cell proliferation and growth, survival and inhibition of apoptosis. KIT and PDGFRA WT GIST, in addition, can harbor mutations in 1 of the 4 subunits of the SDH gene, which cause loss of function. This leads to cytoplasmatic accumulation of succinate, which downregulates prolyl hydroxylase, responsible for promoting HIF1A degradation. Succinate accumulation results in increased levels of HIF1A, which enters the nucleus and activates transcription factors.
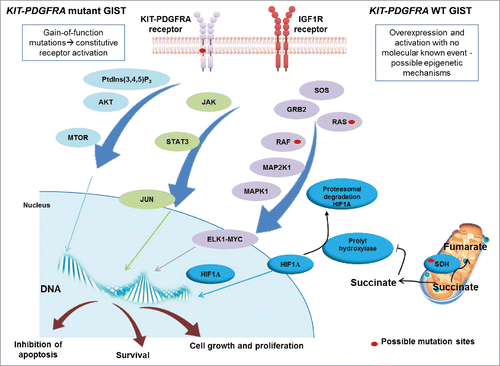
Figure 2. Autophagy as part of the molecular profile in both untreated and imatinib treated GIST cells. In untreated GIST cells (A), autophagy controls tumor cell growth. In treated GIST cells, imatinib binds KIT-PDGFRA receptors, blocking their downstream signaling cascade (B); however, a portion of the cells undergoes quiescence and activates autophagy-related survival mechanisms, which, in turn, may promote growth of resistant subclones, characterized by additional, imatinib-resistant mutations (C).
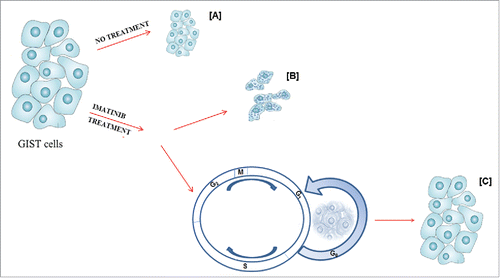
Figure 3. GIST and autophagy signaling. GIST cells can take advantage of autophagy by 2 different mechanisms. In untreated cells, autophagy controls tumor cells growth, whereas, in imatinib-treated cells autophagy is a stress response to starvation. In treated GIST cells, the pro-apoptotic BECN1 (Beclin 1)-PtdIns(3,4,5)P3 complex and LC3-II are expressed at high levels, while the anti-autophagy BECN1-BCL2 complex is at low levels. Autophagy signaling involves 2 key regulators, ATG7 and ATG12, that drive the cells toward autophagy and quiescence. ATG7 and ATG12 inhibition in association with imatinib promotes death cell through apoptosis.
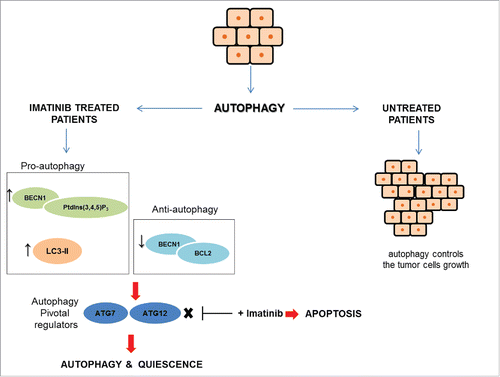
Figure 4. GIST and apoptosis signaling. Survival signals can activate the PtdIns(3,4,5)P3-AKT cascade, which phosphorylates and inactivates the pro-apoptotic BCL2-family member BAD. In GIST, pro-apoptotic proteins, such as BAX, are downregulated and anti-apoptotic regulators are expressed. Anti-apoptotic proteins, including the anti-apoptotic BCL2 family members and inhibitor of apoptosis (IAP) proteins, are regulated by DIABLO/SMAC, which is mutated in GIST samples. IAPs are upregulated in GIST. XIAP/BIRC4, directly inhibits effector caspases, whereas BIRC5/survivin has indirect anti-apoptotic effects by stabilizing XIAP and inhibiting DIABLO. Apoptosis is also regulated through the mitochondrial (intrinsic) pathway. Pro-apoptotic BCL2 family proteins, including, BAX and BCL2L11/BIM, are important mediators of these signals. Activation of mitochondria promotes the release of CYCS/cytochrome C that binds APAF1 to form the apoptosome and subsequent activation of the initiator proCASP9. APAF1 interacts with the pro-apoptotic tumor suppressor FAM96A, which is downregulated in GIST.
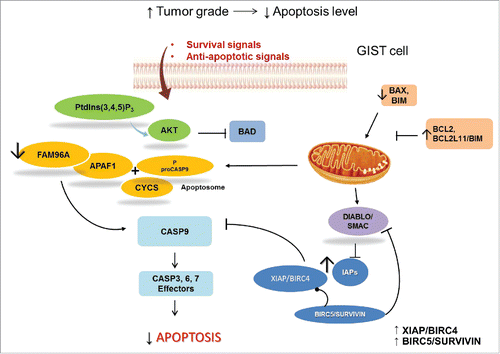
Table 1. Novel therapeutic targets and imatinib combination treatments for GIST.