Figures & data
Figure 1. ATG3 is degraded in response to treatment with DNA-damaging drugs. (A) HCT116 cells were treated with DMSO or etoposide (40 μM) for 3 h and then incubated with fresh medium for the indicated time. Western blotting was performed to detect different ATG proteins. (B) HCT116 cells were treated with etoposide at various concentrations for 3 h and then incubated with fresh medium for 48 h. (C, D) Cisplatin (10 μM) (C) or etoposide (40 μM) (D) were introduced into HCT116 or LoVo cells, respectively. Cells were then treated as described in (A). (E, F) HCT116 cells were treated with etoposide (E) or cisplatin (F) as indicated, and then quantitative PCR (qPCR) was used to measure the mRNA levels of ATG3. The data are presented as the mean ± SD (n = 3). NS, no significance. (G) HCT116 cells were treated with etoposide for 3 h and then incubated with fresh medium for 48 h in the presence or absence of CHQ (10 μM) or MG132 (1 μM). Western blotting was performed to detect endogenous ATG3 protein levels. (H) HCT116 cells were transfected with HA-ubiquitin. Cells were treated 24 h after transfection with or without etoposide for 3 h and then incubated in fresh medium for 48 h. MG132 (1 μM) was added 12 h before collection. Cell lysates were immunoprecipitated with an anti-ATG3 antibody and blotted with an anti-ATG3 or anti-HA antibody.
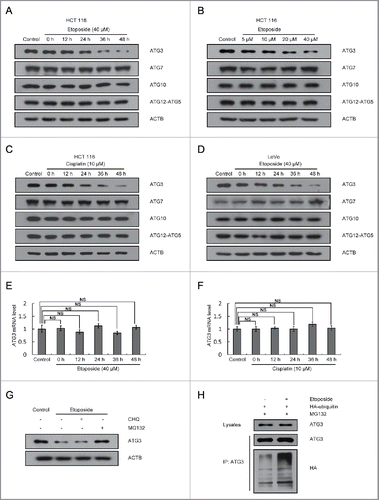
Figure 2. Involvement of PTK2 kinase in ATG3 degradation. (A) MEF cells were treated with or without etoposide (20 μM) for 6 h, and then incubated with fresh medium. The cell lysates were immunoprecipitated with an anti-ATG3 antibody and blotted with an anti-phospho-tyrosine antibody. (B) HCT116 cells were pre-incubated with PF-573228 (PTK2 inhibitor), SRC Inhibitor-1 (SRC inhibitor) or erlotinib (EGFR inhibitor) for 1 h and then treated with or without etoposide (40 μM). Three h later, the medium was changed, and cells were incubated in fresh medium in the presence or absence of kinase inhibitors for the indicated time. Western blotting was performed using anti-ATG3, anti-p-PTK2, anti-p-SRC or anti-p-EGFR antibodies. (C) HCT116 cells were transfected with a PTK2-specific siRNA or negative control. 48 h after transfection, the cells were treated with or without etoposide for 3 h and then incubated with fresh medium for 48 h. Endogenous ATG3 protein levels were measured by western blotting, and the efficiency of PTK2 siRNA was validated by monitoring total PTK2 and p-PTK2. The bands were quantified with ImageJ software. The numbers below the blot indicate the relative levels of p-PTK2 in each group.
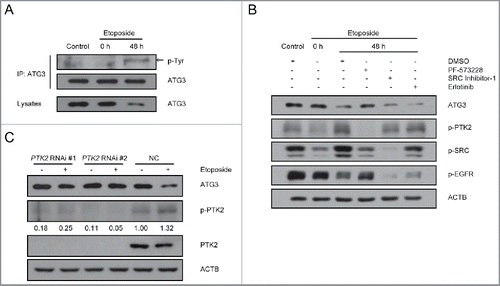
Figure 3. ATG3 degradation is dependent on its Y203 phosphorylation. (A) HCT116 cells were transfected with plasmids encoding FLAG-ATG3 WT or ATG3Y203F. The cell lysates were extracted 24 h after transfection for co-IP with an anti-FLAG antibody and probed with an anti-Y203-phosphorylated ATG3 antibody. (B) MEF cells were treated with or without etoposide (20 μM) for 6 h and then incubated with fresh medium. The cell lysates were immunoprecipitated with an anti-ATG3 antibody and blotted with an anti-Y203-phosphorylated ATG3 antibody. The numbers below the blot indicate the relative levels of p-ATG3 in each group. (C) MEF cells were pre-incubated with PF-573228 for 1 h and then treated with or without etoposide (20 μM) for 6 h. After 48 h incubating in fresh medium, the cells were prepared for co-IP with an anti-ATG3 antibody and detected with an anti-Y203-phosphorylated ATG3 antibody. (D) ATG3-inducible MEF cells were treated with doxycycline (Doxy, 1 μg/ml) to induce WT, Y203E or Y203F ATG3 and then exposed to cycloheximide (CHX, 10 μM/ml). The protein level of ATG3 was measured by western blotting. (E) Graphs show the relative expression of WT or mutant ATG3 with the indicated treatments in (D). The data are presented as the mean ± SD (n = 3). (F) HCT116 cells were co-transfected with FLAG-ATG3 (WT, Y203E or Y203F) and HA-ubiquitin. The cells were treated with MG132 (2 μM) 24 h later for 8 h. Proteins were then immunoprecipitated from the cell extracts with anti-FLAG M2 beads, and the immune-complexes were probed with the anti-ATG3 or anti-HA antibody.
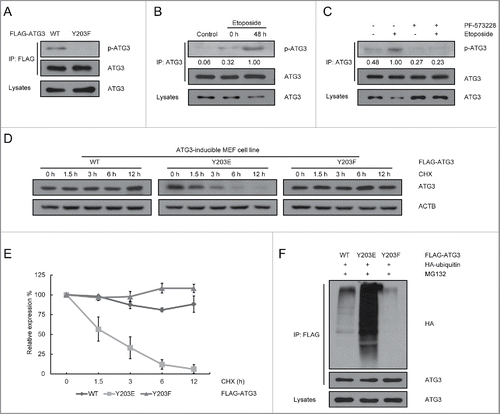
Figure 4. Y203 phosphorylation of ATG3 is not critical for autophagy induction. (A) Atg3 KO MEF cells were transfected with plasmids encoding MYC-his-ATG3 WT or ATG3Y203E. MG132 (2 μM) was added into the medium 12 h after transfection for another 12 h to prevent ATG3Y203E degradation. Cell lysates were extracted to detect ATG3 and MAP1LC3 proteins. (B) Atg3 KO MEF cells were transfected with plasmids encoding FLAG-ATG3 WT, ATG3Y203E or ATG3Y203F. MG132 was added 12 h before collection. Immunofluorescence was performed after staining with anti-FLAG and anti-MAP1LC3 antibodies. Scale bars: 20 μm. (C) Quantification of the MAP1LC3 puncta-positive cells is shown in (B). The criterion for being counted was a cell with more than 10 puncta. The data are presented as the mean ± SD (n = 3). *p < 0.05; NS, no significance. (D) PTK2 stable-knockdown HCT116 cells and WT-HCT116 cells were treated with DMSO or etoposide (40 μM) for 3 h and then incubated with fresh medium for up to 48 h. CHQ (10 μM) was added 1 h before collection. Western blotting was performed to detect ATG3 and MAP1LC3 protein levels. An anti-p-PTK2 antibody was used to validate the efficiency of PTK2 siRNA.
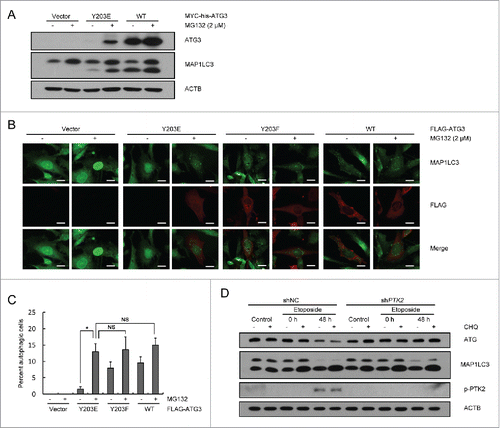
Figure 5. ATG3Y203F is able to promote DNA damage-induced mitotic catastrophe. (A) ATG3-inducible MEF cells were treated with doxycycline (Doxy, 1 μg/ml) to induce WT or Y203F ATG3 and then exposed to etoposide. Then, the cells were stained with Giemsa. Scale bars: 20 μm. (B) Statistical analysis of the abnormal nuclei in (A). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (C) ATG3-inducible MEF cells were treated as described in (A), and immunostained with an antibody against TUBA (green). Nuclei were counterstained with DAPI (blue). Scale bars: 10 μm. (D) Statistical analysis of the abnormal nuclei in (C). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (E) ATG3-inducible MEF cells were treated as described in (A), and the cells were collected and analyzed by FACS. (F) HCT116 cells were treated with DMSO or etoposide (40 μM) for 3 h and then incubated with fresh medium for 48 h. The cell lysates were extracted for co-IP with an anti-ATG3 antibody and probed with an anti-BAG3 antibody.
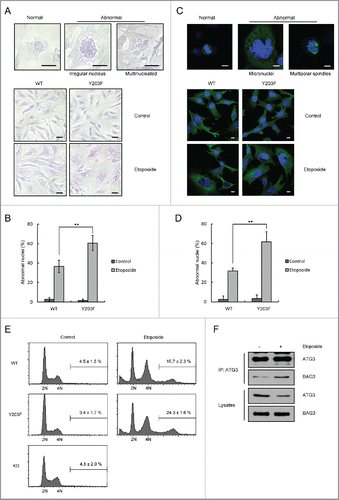
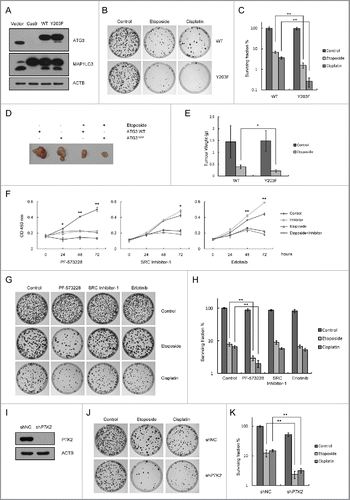
Figure 6. Sustained ATG3 is related to tumor cell growth inhibition in response to DNA-damaging agents. (A) Cas9 ATG3 knockout cells were transfected with plasmids encoding FLAG-ATG3 WT ATG3Y203F. Western blotting was performed to detect the ATG3 and MAP1LC3 protein levels. (B) Cells overexpressing FLAG-ATG3-WT or FLAG-ATG3Y203F from (A) were treated with or without etoposide (40 μM) for 3 h or cisplatin (10 μM) for 6 h. Then, the cells were washed with fresh medium and allowed to grow for approximately 2 wk. The cells were stained with methylene blue before counting. (C) Statistical analysis of the surviving fraction in (B). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (D) Cells overexpressing FLAG-ATG3-WT or FLAG-ATGY203F cells were injected into the NOD-SCID mice, and the tumors were detected 3 weeks later (4 mice per group). (E) Tumors were weighed and analyzed. *p < 0.05. (F) HCT116 cells were pre-incubated with PF-573228, SRC Inhibitor-1 or erlotinib for 1 h and then treated with or without etoposide (40 μM) for 3 h. Cell proliferation was tested with the WST-1 assay, and the data are presented as the mean ± SD. Student t test was performed by comparing drug-treated cells versus untreated cells. *p < 0.05; **p < 0.01. (G) HCT116 cells were pre-incubated with PF-573228, SRC Inhibitor-1 or erlotinib for 1 h and then treated with or without etoposide or cisplatin. Then, the cells were treated as described in (B). (H) Statistical analysis of the surviving fraction in (G). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01. (I) Western blotting was performed to detect the endogenous PTK2 level in stable PTK2 knockdown and negative control cell lines. (J) PTK2 stable-knockdown HCT116 cells and WT-HCT116 cells were treated with or without etoposide or cisplatin. Then, the drug was removed and the colonies were counted after 2 wk. (K) Quantification of the surviving fraction in (J). The data are presented as the mean ± SD based on 3 independent experiments. **p < 0.01.