Figures & data
Figure 1. Altered ultrastructure of ATP6AP2-depleted liver. (A) Schematic illustration of the v-H+-ATPase complex composition. The v-H+-ATPase V1 sector (yellow and brown subunits, upper case letters) transfers energy released by ATP hydrolysis into a rotational force that triggers the transport of protons through the V0 sector (blue subunits, lower case letters). ATP6AP2 is an accessory subunit that acts as a chaperone for V0 sector assembly. (B) Livers from Atp6ap2 cKO mice weigh more than those of wild-type (WT) animals. (C) Immunoblot analysis of liver lysates confirms reduced concentrations of ATP6AP2 full length (fl) and C-terminal fragments (CTF) in ATP6AP2-depleted samples that come with lower v-H+-ATPase V0 sector concentrations. GAPDH detection was used as a control for equal protein load. (D) Transmission electron micrographs reveal ultrastructural changes in Atp6ap2 cKO liver as compared with wild-type samples. Affected hepatocytes (Hep) accumulate vesicles (arrows) that contain partially degraded cytoplasmic material. The boxed areas are resolved with higher magnification, Nu, nucleus.
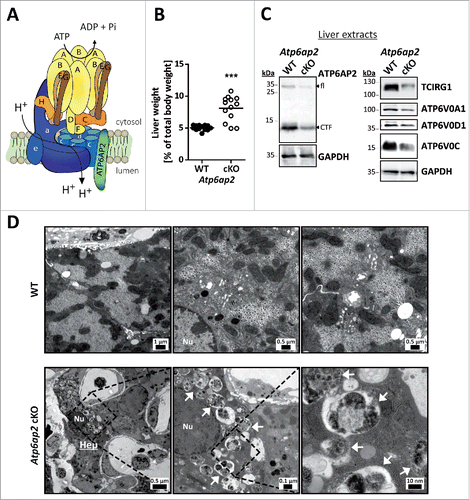
Figure 2. Accumulation of autophagic vacuoles and lysosomes in Atp6ap2 conditional knockout liver. (A) Immunofluorescence analyses of liver sections show an increase in LC3-, SQSTM1- and LAMP1-positive vesicles in ATP6AP2-depleted hepatocytes (cKO) in comparison to control tissue (WT). Scale bars: 10 µm. (Bi) Immunoblot analysis of autophagic marker proteins LC3-I, LC3-II and SQSTM1 and of lysosomal LAMP1 in liver lysates (15 μg per lane) from Atp6ap2 cKO and wild-type mice. All protein concentrations were increased in the knockout samples. (Bii-iv) Quantification of the immunoblots was carried out in relation to GAPDH levels. Shown are mean values ± standard errors from 6 to 7 independent animal preparations per genotype. **P < 0.01, ***P < 0.001 according to unpaired, 2-tailed Student t test.
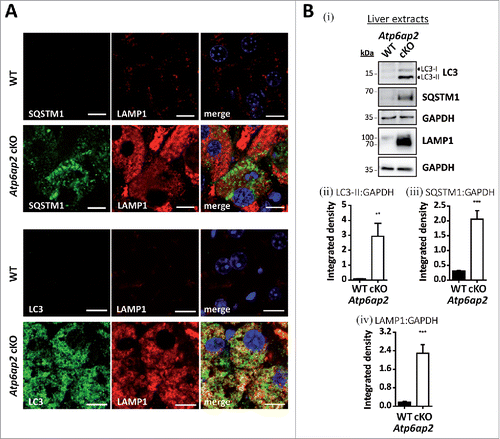
Figure 3. Activation of MTORC1 and lysosomal MTOR recruitment are late responses in ATP6AP2-depleted liver. (A) Time course of the expression levels of selected proteins in wild-type and Atp6ap2 conditional knockout (cKO) liver following the first application of poly (I:C) at d 1. Phosphorylated (p-) and total protein were detected for the MTORC1 targets RPS6KB (Thr389) and ULK1 (Ser757) as well as of the MTORC1 repressor AKT1S1 (Thr246). Detection of GAPDH was used to visualize equal protein loading. * indicates nonspecific antibody binding. CTF, C-terminal fragments. (B) Fluorescence microscopy analysis of MTOR localization in fixed wild-type and Atp6ap2 cKO hepatocytes (d 10). A co-staining of LAMP2 and TCIRG1/ATP6V0A3 (V0 a3) was used to assess lysosomes and the Atp6ap2 knockout efficiency, respectively. Boxed areas are resolved with higher magnification. Scale bars: 10 µm. (C) Liver lysosomes (tritosomes) were isolated from 2 wild-type and 2 Atp6ap2 cKO mice (d 15) and processed for immunoblotting. Atp6ap2 cKO tritosome preparations display increased levels of the kinase MTOR as well as of the MTORC1 recruiting proteins RRAGA, RRAGC, LAMTOR1 and LAMTOR2. LAMP2 levels in the isolated lysosomes were not changed between the genotypes. The experiment was reproduced using a second set of 2 wild-type and 2 Atp6ap2 cKO tritosome preparations (not shown).
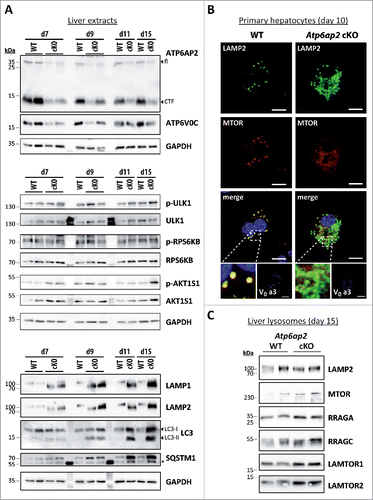
Figure 4. MTORC1 is active in liver bearing decreased v-H+-ATPase levels. (A) Phosphorylation of the MTORC1 targets RPS6KB and ULK1 as well as of the MTORC1 repressor AKT1S1 and the insulin-responsive kinases MAPK1/ERK2-MAPK3/ERK1 (Thr202, Tyr204) and AKT (Ser473) was analyzed by immunoblotting of 4 different wild-type control (WT, lanes 1–4) and 4 Atp6ap2 cKO liver extracts (lanes 5–8) and revealed increased signaling activity in the Atp6ap2 cKO samples. GAPDH staining was used to control for equal protein loads. Irrelevant bands according to the antibody datasheet are labeled with *. (B) Signal intensities from (A) were quantified in regard to the extent of protein phosphorylation and compared between both genotypes. Shown are means ± standard errors from 4 independent liver preparations per genotype. * P < 0.05, ** P < 0.01 according to unpaired, 2-tailed Student t test. (C) Transcription of TFEB-dependent and -independent genes involved in autophagy and lysosomal biogenesis was analyzed by qRT-PCR using whole liver RNA extracts of wild-type and Atp6ap2 cKO animals. Expression levels were normalized to the most stable housekeeping genes and related to the wild-type expression rate. Bars represent mean values of 6 independent RNA preparations per genotype ± standard errors (** P < 0.01 according to unpaired, 2-tailed Student t test between genotypes). (Di) Fractionation of cytosolic (cyt) and nuclear (nuc) proteins from wild-type and Atp6ap2 cKO livers. TFEB displays prominent nuclear localization in both genotypes. SP1 and GAPDH were used as specific nuclear or cytosolic positive control, respectively. (Dii) Quantification of data obtained in (Di) regarding the ratio of nuclear to cytosolic TFEB. Shown are mean values ± standard errors of 6 independent preparations per genotype.
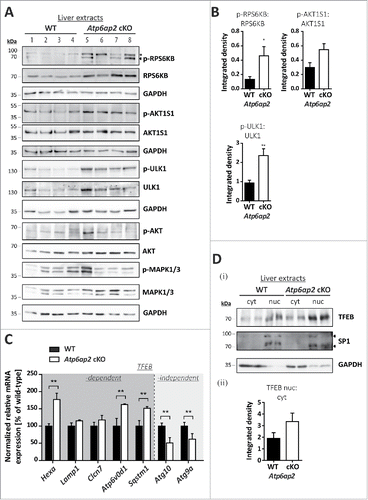
Figure 5. Reduced v-H+-ATPase concentration does not impair autophagic degradation in Atp6ap2 knockout MEFs. (A) Lysates of Atp6ap2 knockout (KO) mouse embryonic fibroblasts lack ATP6AP2 full length (fl) and C-terminal fragments (CTF) in immunoblot analyses. Concomitant probing for the indicated v-H+-ATPase V0 and V1 subunits revealed reduced V0 sector level upon deletion of Atp6ap2. ACTB served as a loading control. (B) Immunofluorescence staining of control and ATP6AP2-deficient MEFs confirmed reduced levels of TCIRG1/ATP6V0A3 that shows a colocalization with the lysosomal marker LAMP2 in wild-type cells. Scale bar: 10 µm. (C) Fibroblasts of both genotypes were cultivated in the presence or absence of free amino acids (AA) and serum for 2 h. Where indicated, cells were pre-treated with 100 nM BafA1 for 1 h to inhibit lysosomal degradation and the treatment continued throughout the experimental setup. Control fibroblasts were incubated in the presence of DMSO (i). Levels of LC3-I, LC3-II and SQSTM1 were detected by immunoblot analyses. Labeling of GAPDH was used to control for an equal protein load. (ii, iii) Quantification of (i) revealed a starvation-induced elevation in the ratio LC3-II to GAPDH and a parallel decrease in the amount of SQSTM1 that appeared independent of the expression of ATP6AP2. Shown are means ± standard errors from 4 independent measurements.
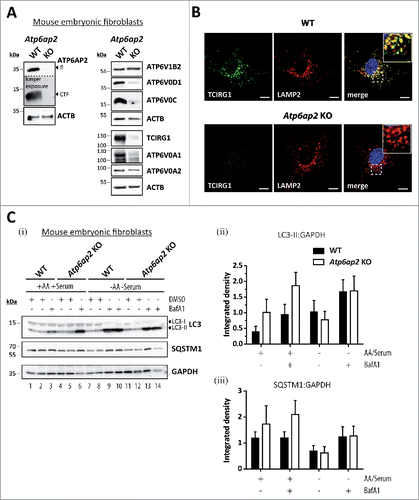
Figure 6. Reduction of the v-H+-ATPase V0 sector level interferes with amino acid-dependent activation of MTORC1. (A) Wild-type control and Atp6ap2 knockout MEFs were cultivated for 1 h in the absence of amino acids and serum to silence MTORC1 signaling. Then, the MTORC1 complex was activated in amino acid-containing DMEM for up to 2 h. Simultaneous treatment with Torin 1 (250 nM) was used to block MTORC1 activation. Basal MTORC1 activity was assessed under fed conditions (untreated). (i) Detection of the total and phosphorylated MTORC1 substrate RPS6KB and AKT1S1 in control and ATP6AP2-deficient MEFs following the stimulation with amino acids. TFEB phosphorylation correlated with the activation of RPS6KB and was decreased in amino acid-stimulated Atp6ap2 knockout cells. GAPDH and EEF2 served as loading controls. (ii) Ratios of p-RPS6KB to total RPS6KB were quantified from 3 independent experiments and are shown as means ± standard errors. (B) Reactivation of MTORC1 by extracellular application of BSA. Fibroblasts of both genotypes were starved for amino acids and serum for 1 h before cells were incubated with 3% (w:v) BSA for the indicated time periods to reactivate MTORC1 following intracellular digestion. (i) Addition of BSA was sufficient to trigger phosphorylation of RPS6KB as well as AKT1S1 regardless of the ATP6AP2 expression. Control samples were left untreated or incubated with the MTOR inhibitor Torin 1. (ii) Quantification of the signal intensities of p-RPS6KB to total RPS6KB depicted as means ± standard errors from 3 independent experiments.
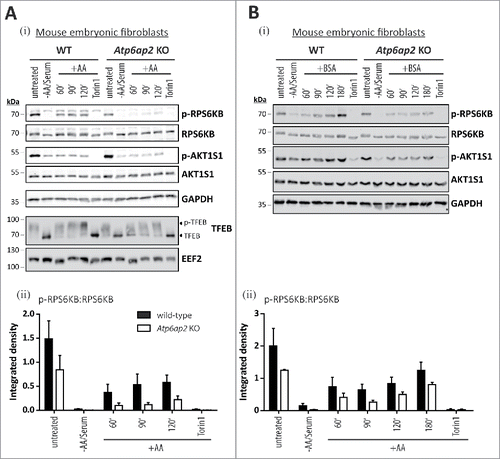
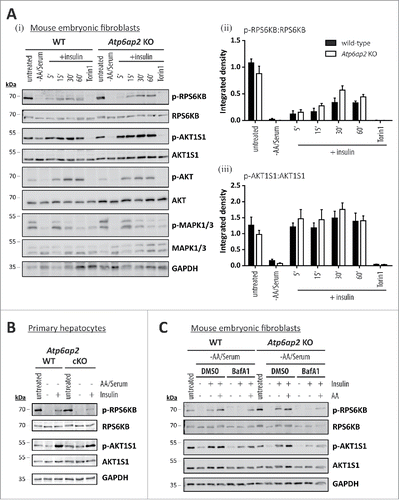
Figure 7. Activation of MTORC1 by insulin depends on amino acids but not on ATP6AP2 expression. (A) Activation of MTORC1 was triggered in wild-type control (WT) and Atp6ap2 knockout (KO) MEFs by 1 h starvation of serum and amino acids and subsequent administration of insulin (150 nM) for 5 to 60 min. Control samples were cultivated in the presence of serum and amino acids (untreated) or co-cultivated with insulin and Torin 1 (250 nM) to interfere with MTORC1 stimulation. (i) Insulin treatment led to elevated signals for phosphorylated AKT1S1 (Thr246) in both genotypes and a concomitant increase in RPS6KB phosphorylation (Thr389). Phosphorylated and total MAPK1/ERK2-MAPK3/ERK1 as well as AKT were assessed to verify insulin receptor activation. Mean ratios of p-RPS6KB to RPS6KB (ii) and p-AKT1S1 to AKT1S1 (iii) as detected in (i) were quantified from 3 independent experiments. Error bars display standard errors. (B) Insulin-dependent activation of MTORC1 in primary wild-type and Atp6ap2 conditional knockout (cKO) hepatocytes as described above (A). Cells were starved for amino acids and serum for 1 h before MTORC1 activity was triggered by application of 150 nM insulin for 15 min. (C) Control and Atp6ap2 knockout fibroblast were maintained in the presence of 50 nM BafA1 for 16 h to block lysosomal proteolysis or autophagy, respectively. MTORC1 activation was then triggered by starvation and stimulation with insulin for 15 min as described in (A), but under continued inhibitor treatment. Simultaneous reactivation of MTORC1 by amino acids and insulin was included to replenish intracellular amino acid pools. Cells were kept in DMEM containing serum and amino acids (untreated) as a control for the steady-state activity of MTORC1 or processed with DMSO as solvent control.