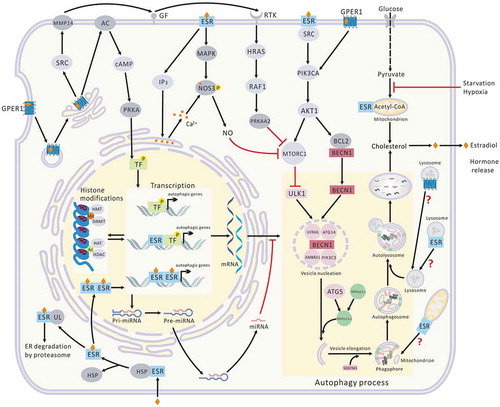
Figures & data
Table 1. The regulation of E2 on autophagy.
Table 2. The regulation of autophagy by ESR ligands a.
Table 3. Autophagy proteins regulated by E2 via TFs.