
Figures & data
Figure 1. The biogenesis of LDs. The process of de novo LD biogenesis can be divided in three main discrete steps: (A) Nucleation, (B) growth and (C) budding. The nucleation step is characterized by the formation of an oil lens structure in between the two lipid bilayers of the ER limiting membrane, which is catalyzed by ER resident proteins such as FITM1/FITM2 and BSCL2/seipin. The growth of the nascent LDs starts with an accumulation of TAGs and SE, and involves a ripening phenomenon that regulates their size. The partial (in yeast) or complete (in mammalian cells) detachment of the LDs from the ER membrane defines the LDs budding. LDs can also expand (D) by increasing their size through the coalescence of LDs and/or the local synthesis of TAGs. Several enzymes involved in the biogenesis and function of LDs, which are discussed in the review, are indicated.
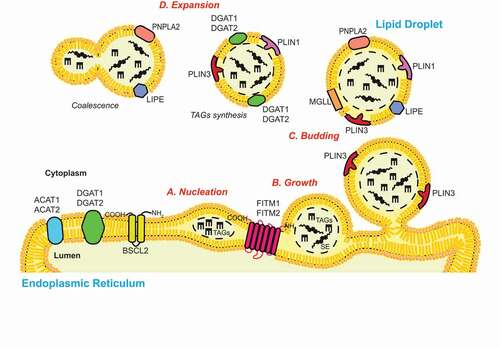
Figure 2. Autophagic processes of LDs. (A) Macrolipophagy involves the sequestration of LDs by autophagosomes and their subsequent delivery to lysosomes/vacuoles for turnover. LDs could be selectively recognized in both an Ub-dependent and -independent manner. On the one hand, during Ub-dependent macrolipophagy, the LDs surface protein AUP1 binds and recruits the Ub conjugating enzyme UBE2G2, which could represent the functional link to the subsequent Ub-dependent macroautophagic degradation of LDs. On the other hand, SPART binds the LDs-anchored protein PLIN3 and promotes the recruitment and activation of the Ub ligase ITCH/AIP4. ITCH polyubiquitinates PLIN2 (and potentially other LDs-associated proteins), leading to the association of the macroautophagy cargo receptor SQSTM1/p62, which triggers macrolipophagy. During Ub-independent microlipophagy, both PNPLA2/ATGL and LIPE/HSL, which are distributed on the phospholipid monolayer limiting LDs, interact with MAP1LC3A/LC3 via their LIR motifs, inducing the local, in situ, formation of a phagophore. It remains unknown whether the Ub-dependent and – independent macrolipophagy are mutually exclusive or act concomitantly/sequentially. RAB10, RAB7, EHBP1 and EHD2 have also been implicated in macrolipophagy. However, it is unclear whether they are playing a role in the Ub-dependent, the Ub–independent or both processes of macrolipophagy. (B) Microlipophagy has been better characterized in yeast S. cerevisiae. It involves the direct engulfment of LDs by vacuoles. Microlipophagy relies on liquid ordered and sterol enriched vacuolar microdomain formation. The sterol-transporting proteins Ncr1 and Npc2 are essential for the formation of these vacuolar microdomains and subsequent LD engulfment. The formation of these vacuolar microdomains also involve core ATG proteins such as Atg7, Atg8, Vps30/Atg6 and Atg14. Additionally, an ATG core machinery-independent microlipophagy has been described and requires ESCRT components such as Vps24 and Vps27 (C) During CMA of LDs-anchored proteins, the cytosolic chaperone HSPA8/HSC70, together with its co-chaperones, recognizes proteins possessing a KFERQ motif, including PLIN2, PLIN3 and PLIN5. This cargo-chaperone complex then binds LAMP2A present of the surface of lysosomes, which mediates the unfolding and translocation into the lumen of this organelle of the targeted cargo to be degraded. CMA-mediated degradation of PLIN proteins promotes the recruitment of PNPLA2/ATGL and ATG machinery components onto LDs, making CMA an upstream regulator of both neutral lipolysis and macrolipophagy.
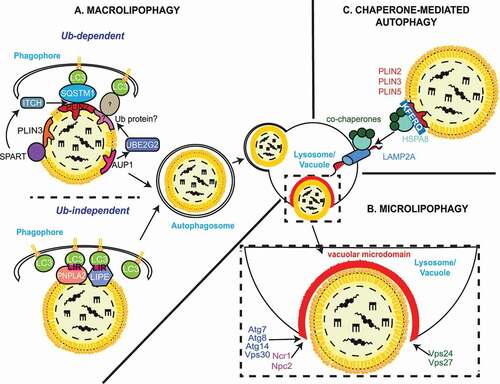
Figure 3. Dysregulated lipid metabolism plays a role in the development of NASH. Altered lipophagy, lipolysis and de novo lipogenesis in the liver contribute to NASH development through fat accumulation, and subsequent inflammation and fibrosis. A “first hit” of increased FFAs leads to accumulation of LDs in hepatocytes, a condition known as NAFLD. Additional “hits”, including inflammation and insulin resistance contribute to steatohepatitis (NASH), characterized by hepatocyte ballooning, lobular inflammation and fibrosis. Liver-resident macrophages (KCs) may acquire a pro-inflammatory phenotype due to the excess hepatic fat burden and mediate the inflammatory response through the recruitment of immune cells from the periphery. In response to hepatic injury, normally quiescent HSCs become activated by various signals. Enhanced lipophagy of vitamin A-storing LDs provides energy for HSCs activation, leading to hepatic fibrosis through HSCs-mediated collagen deposition.
Table 1. Mouse models of NASH