Figures & data
Figure 1. Dual role of copper in cancer development. On the one hand, elevated copper levels can promote tumor growth by inducing ROS production, exacerbating genomic instability, and affecting various tumor-associated signal transduction events. On the other hand, excessive copper concentrations can induce tumor cell death when they exceed a certain threshold limit.
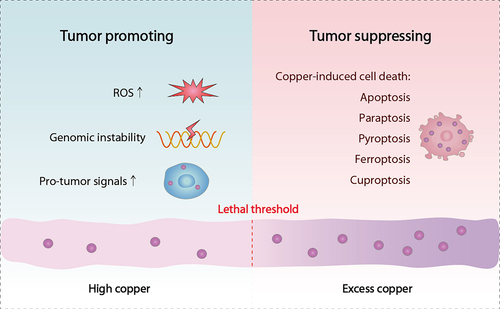
Figure 2. Molecular mechanisms of copper metabolism. Copper metabolism is a complex dynamic process regulated at the cellular and organ level by multiple molecules. The uptake of copper ions is mediated by SLC31A1 and SLC31A2, while the export of copper is driven by ATP7A and ATP7B. In cells, copper is transported to different subcellular organelles for bioavailability by several copper-binding proteins, including COX17, CCS, and ATOX1. Furthermore, the binding of MT1, MT2 and GSH to copper can limit the cytotoxicity of copper excess.
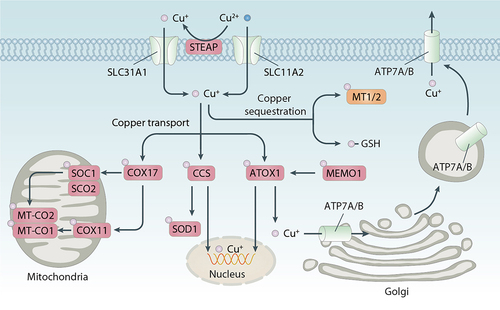
Figure 3. Types of regulated cell death. Regulated cell death is a biologically controlled process involved in various physiological or pathological events. It can be divided into apoptotic and non-apoptotic cell death. Compared with apoptosis, which generally requires the activation of caspase proteases, non-apoptotic cells are mostly caspase-independent and have the morphological characteristics of necrosis.
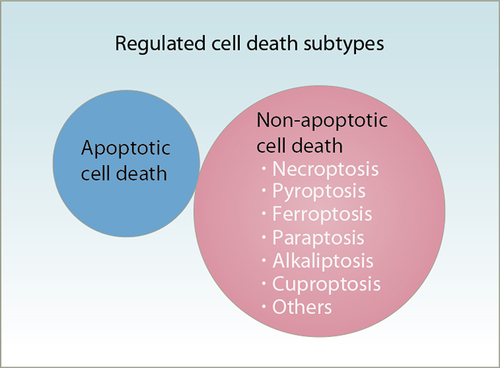
Figure 4. Role of copper in apoptosis. (A) Mechanism of copper-induced apoptosis. Copper induces apoptosis primarily through the induction of ROS, DNA damage, and proteasome inhibition. The apoptotic process is initiated by mitochondria-intrinsic apoptotic signals. CYCS-activated CASP9 propagates the apoptotic cascade by activating downstream CASP3, a key apoptosis execution protein that participates in the cleavage of multiple substrates. Furthermore, AIFM1 is released from mitochondria and induces caspase-independent apoptosis by attacking DNA. (B) the role of TP53 in copper-induced apoptosis. Copper induces TP53-dependent apoptosis by activating transcription of TP53 target genes, including BAX, CDKN1A/p21, PMAIP1/NOXA, and BBC3/PUMA. Copper also induces TP53-independent apoptosis by inhibiting ribosome synthesis and inducing nucleolar stress. (C, D) the anti-apoptosis role of copper. IL17 released by immune cells can increase STEAP4-mediated intracellular copper levels, leading to 5-fluorouracil (5-FU) resistance by activating the anti-apoptotic XIAP protein. Copper triggers the upregulation of CD274/PD-L1, which induces tumor immune escape by binding with PDCD1/PD-1 on activated T cells.
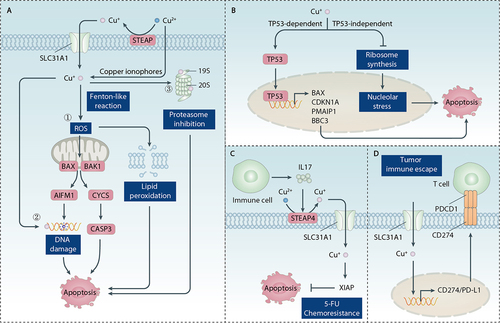
Figure 5. Role of copper in paraptosis. Paraptosis is a form of regulated necrosis characterized by vacuolation of mitochondria or ER. Copper promotes paraptosis by inducing proteasome inhibition, ER stress, Ca2+ imblance, and ROS production.
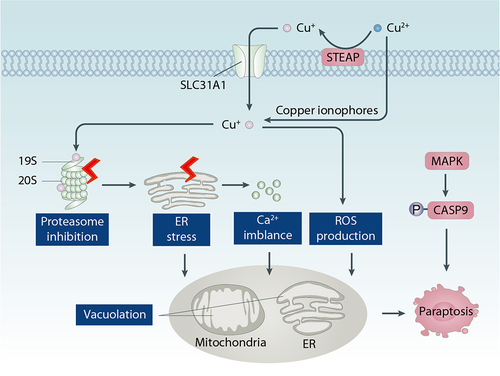
Figure 6. Role of copper in pyroptosis. Copper promotes pyroptosis by inducing ROS production and ER stress, which leads to the formation of the NLRP3 inflammasome and the creation of membrane pores through the action of GSDMD.
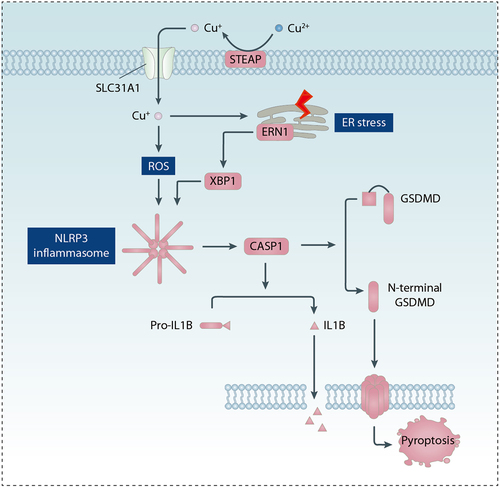
Figure 7. Role of copper in ferroptosis. (A) Pro-ferroptotic role of copper. Copper increases intracellular ROS through Fenton-like reactions or mitochondrial damage. Subsequently, increased ROS lead to lipid peroxidation of the plasma membrane or membrane structures. Furthermore, copper directly binds to GPX4 and induces GPX4 oligomerization, ultimately promoting the autophagic degradation of GPX4 mediated by the autophagy receptor TAXIBP1. (B) Anti-ferroptotic role of copper. COMMD10 is a key copper metabolism protein that reduces intracellular copper levels. Copper promotes HIF1A stabilization, thereby increasing transcription of HIF1A target genes, including FABP3, FABP7, CP, and SLC7A11. Copper-mediated upregulation of these HIF1A target genes suppresses lipid peroxidation and ferroptosis.
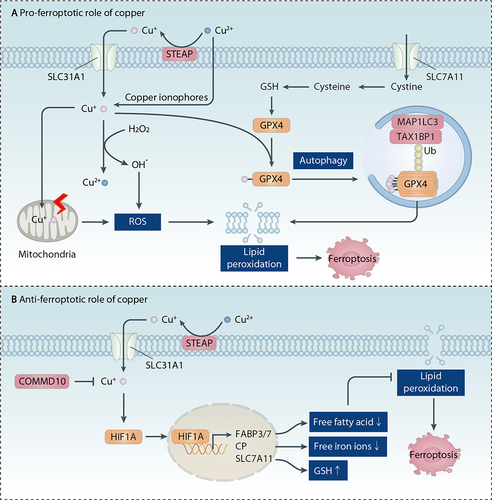
Figure 8. Role of copper in cuproptosis. Cuproptosis is induced by enrichment of copper through SLC31A1 uptake or copper ionophores. ATP7A and ATP7B have copper efflux functions, thereby inhibting cuproptotic cell death. The cell death process is related to mitochondrial proteotoxic stress caused by lipoylation of subunites of the PDH complex, such as DLAT. FDX1 plays a key role in reducing Cu(II) to the toxic form, Cu(I), increasing DLAT protein lipidation, and promoting Fe-S cluster protein loss. Furthermore, copper-mediated damage to the mitochondrial respiratory chain causes hyperactivation of the energy sensor AMPK, which accelerates cuproptosis and the release of the pro-inflammatory mediator HMGB1.
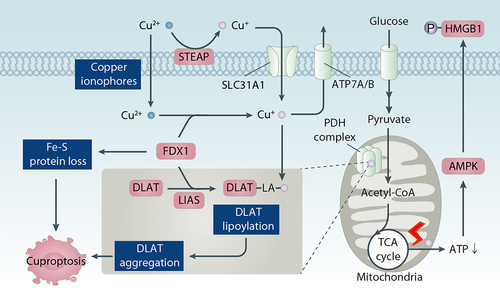
Figure 9. Role of copper in autophagy. Autophagy is a lysosome-mediated degradation process characterized by the formation of multiple member structures such as phagohpores, autophagosomes, and autolysosomes. Copper can induce autophagy initiation by activating AMPK or inhibiting the MTOR kinase pathway or directly binding to ULK1 or ULK2 kinase. Copper-mediated increase in MAP1LC3 and activation of TFEB transcription factor contribute to the formation of autophagosomes and autolysosomes, respectively. Functionally, copper-induced autophagy can lead to a protective response and autophagic cell death, respectively, depending on the strength of the stimulus and the type of substrate being degraded.
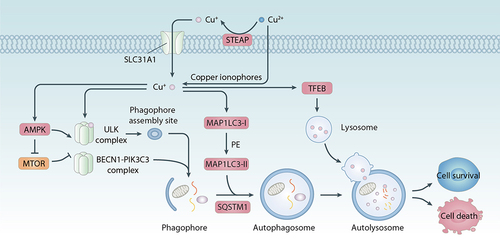
Figure 10. Copper chelators and ionophores in cancer therapy. Because copper supports the rapid growth of cancer cells, copper chelators can inhibit the functions of copper and induce apoptosis or necrosis of cancer cells. In contrast, copper ionophores transport copper into cells and induce copper overload and cytotoxicity. Copper-mediated cell death, including apoptosis, paraptosis, ferroptosis, and cuproptosis, plays a context-dependent role in tumor therapy.
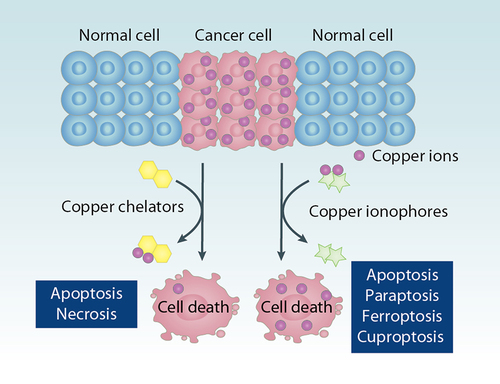
Table 1. Copper chelators for cancer treatment.
Table 2. Copper ionophores for cancer treatment.
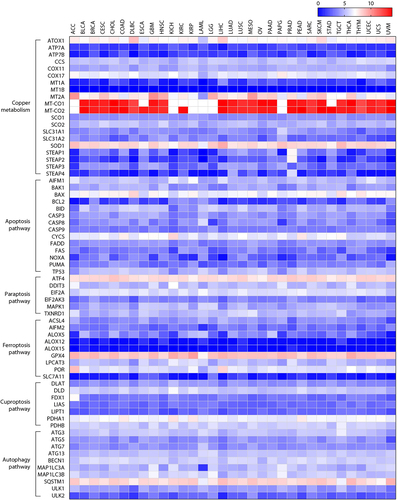
Figure 11. Gene expression changes in cancer patients. The gene expression of key regulators of copper metabolism, cell death, and autophagy pathway in patients with cancer was analyzed using the web server GEPIA (http://gepia.cancer-pku.cn/). Log 2 (TPM+1) was used for log-scale. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B‐cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma.