Figures & data
Figure 1. Differences among GPX family members. Briefly, human GPX members can be divided into groups of selenoproteins (GPX1, 2, 3, 4, and 6) and non-selenoproteins (GPX5, 7, and 8) with different subcellular locations, spatial structures, and molecular masses.
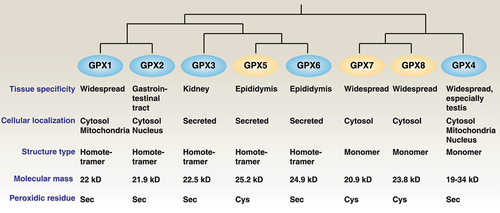
Table 1. Developmental and disease phenotypes associated with GPX4 deficiency in mice.
Figure 2. GPX4 isoenzymes. The GPX4 protein has three isoforms (cGPX4, mGPX4, and nGPX4) encoded by distinct 1a and 1b exons. They exhibit overlapping and distinct functions in regulating development and cell death.
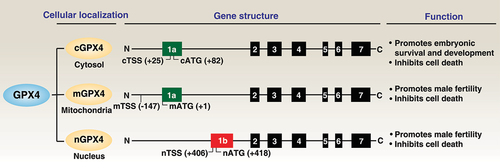
Figure 3. Role of GPX4 in lipid peroxidation. Polyunsaturated fatty acids (PUFAs) are major components of cell membrane phospholipid bilayers and are susceptible to oxidative damage that produces the toxic lipid oxidation product PLOOH. In contrast, membrane system xc− is a heterodimeric complex composed of SLC7A11 and SLC3A2, which mediates cystine uptake into cells that is followed by GSH synthesis via the GCLC-GSS pathway. GSH is a co-factor in the catalytic cycle of GPX4 that detoxifies PLOOH to the lipid alcohol PLOH. Therefore, the pharmacological inhibition of GCLC, SLC7A11, and GPX4 using indicated small-molecular compounds can promote or enhance lipid peroxidation.
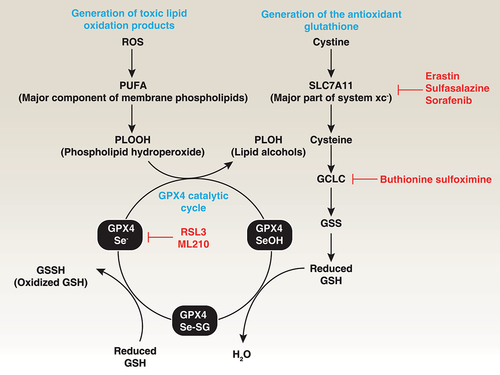
Figure 4. Expression and modification of GPX4. (A) Multiple transcription factors are required for GPX4 expression. As a selenoprotein, Sec-tRNA[Ser]Sec requires a Sec insertion sequence element in Gpx4 mRNA. (B) GPX4 can undergo ubiquitination (Ub), phosphorylation (P), sumoylation (Su) and alkylation (Alk) at specific sites. (C) Both the UPS and autophagy are involved in GPX4 protein degradation.
![Figure 4. Expression and modification of GPX4. (A) Multiple transcription factors are required for GPX4 expression. As a selenoprotein, Sec-tRNA[Ser]Sec requires a Sec insertion sequence element in Gpx4 mRNA. (B) GPX4 can undergo ubiquitination (Ub), phosphorylation (P), sumoylation (Su) and alkylation (Alk) at specific sites. (C) Both the UPS and autophagy are involved in GPX4 protein degradation.](/cms/asset/20575311-76f4-45d6-8536-9688850c856a/kaup_a_2218764_f0004_oc.jpg)
Figure 5. GPX4 and cell death. Regulated cell death mechanisms have been studied in a wide range of oxidative stress models. The overexpression of GPX4 blocks cell death, whereas GPX4 depletion induces cell death in a cell-type and stimulus-dependent manner. For example, GPX4 has been demonstrated to be a negative regulator of apoptosis (A), necroptosis (B), pyroptosis (C), ferroptosis (D), or parthanatos (E).
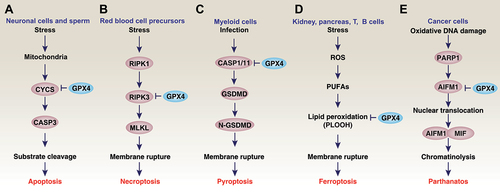
Figure 6. GPX4 and autophagy. Autophagy is a dynamic degradation process that involves the formation of multiple membrane structures. In addition to the classical autophagic machinery, the induction of autophagy-dependent ferroptosis requires specific proteins, such as TMEM164, which is responsible for the formation of phagophores for autophagosome generation. The selective degradation of pro-survival proteins or organelles by autophagy promotes ferroptosis, including the passive release of DAMPs. On the other hand, the early secretion of inflammatory mediators such as DCN during ferroptosis is involved in secretory autophagy mediated by the fusion of the plasma membrane. The autophagic degradation of GPX4 is mediated by different mechanisms. For example, copper and erastin can promote GPX4 degradation by using autophagic receptors TAXIBP1 and SQSTM1, respectively. In addition to macroautophagy, CMA also mediates GPX4 degradation in response to erastin. This process can be inhibited by phosphorylated CKB at T133, which leads to GPX4 phosphorylation at S104.
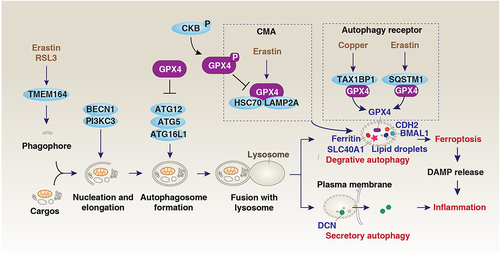
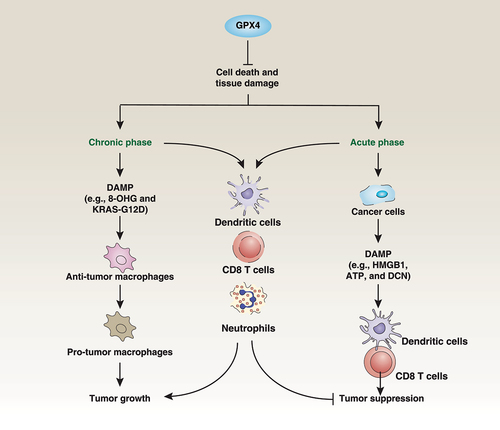
Figure 7. Dual roles of GPX4 in cancer. The loss of GPX4 results in cell death and tissue damage. On the one hand, chronic inflammation may occur, leading to DAMP-mediated macrophage polarization in the tumor microenvironment, which can promote tumor growth. On the other hand, acute cell death caused by the loss of GPX4 can clear tumor cells and release DAMPs, activating anti-tumor immunity. GPX4 is widely expressed in cancer cells and immune cells, including DCs, CD8 T cells, and neutrophils. However, current GPX4 inhibitors are nonselective and can also cause immune cell death, leading to inhibition of anti-tumor immunity.