
Figures & data
Figure 1. Molecular patterns from intestinal inflammation to CAC development. The initial phase of intestinal inflammation activates the NFKB (nuclear factor kappa B) in immune cells via TNF (tumor necrosis factor), IL1, and danger-associated molecular patterns (DAMPs), which leads to an influx of pro-inflammatory factors, including TNF, IL1, IL6, IL11, IL22, VEGF (vascular endothelial growth factor), ROS, among others. TNF plays a pivotal role by activating fibroclasts to produce VEGF, which provides essential nutritional support for tumor growth, and concurrently stimulates Th17 cells to produce IL17 and IL22. In the presence of ROS and RNS, further DNA damage is induced within cells, a crucial event in the progression of CAC. This DNA damage, along with the concerted activation of STAT and NFKB within tumor cells by cytokines, ultimately drives tumor proliferation.
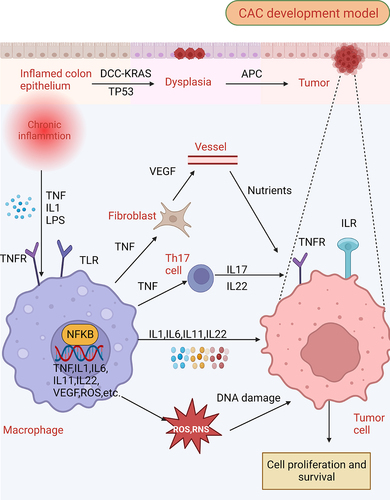
Figure 2. Defective autophagy leads to an increase in intestinal inflammation. (A) autophagy promotes the membrane localization of OCLN (occludin), a barrier-forming protein, by reducing its endocytosis in an MAPK1/ERK2-MAPK3/ERK1-dependent manner. In contrast, defective autophagy fails to degrade the pore-forming protein CLDN2, leading to increased intestinal permeability and bacterial translocation. (B) autophagy-deficient intestinal epithelium cells exhibit increased apoptosis and an increased generation of danger/damage-associated molecular patterns (DAMPs) and promotes the generation of inflammation. (C) during an invasive pathogen infection, lysozyme is secreted by the secretory autophagy pathway. This process requires endoplasmic reticulum (ER) stress-mediated EIF2AK3-EIF2A activation in Paneth cells. Contrastingly, impaired autophagy leads to the impaired ability of Paneth cells to secrete lysozyme, further increasing pathogen invasion. (D) damage of ATG16L1 in intestinal stem cells can lead to failed cell protection induced by muramyl dipeptide (MDP) and damage to the homeostasis of intestinal stem cells. Moreover, autophagy defects lead to the increased sensitivity of differentiated cells to reactive oxygen species (ROS) produced by commensal bacteria and may inactivate the hpo-yki pathway to promote the excessive proliferation of stem cells.
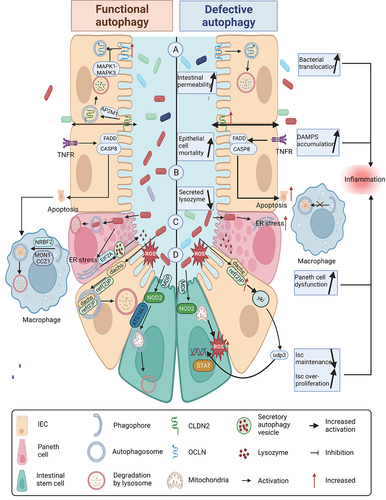
Figure 3. Autophagy deficiency promotes CAC initiation. (A) defects in autophagy lead to elevated levels of reactive oxygen species (ROS) and endoplasmic reticulum (ER) stress. The increased ROS can induce DNA damage in epithelial cells, while ERN1/IRE1α (endoplasmic reticulum to nucleus signaling 1) elevation promotes the production of CTNNB1. PDIA2 (protein disulfide isomerase family a member 2) further facilitates metabolic reprogramming. (B) during the impairment of autophagy, obstacles to the activation of inflammasomes are eliminated, which promotes the cleavage of GSDMD (gasdermin D) to form N-GSDMD (N-terminal GSDMD). This process enables the conversion of the IL1B precursor into its mature form, and stimulates the production of matrix metalloproteinases (MMPs). (C) defects in autophagy contribute to an increase in invasive pathogenic microorganisms such as adherent-invasive E. coli (AIEC) and colibactin-producing E. coli (CoPEC), leading to the production of inflammatory mediators such as TNF and IL6. CoPEC can directly induce DNA damage in epithelial cells. Moreover, autophagy defects increase ER stress and decrease akkermansia muciniphila abundance, thereby reducing the proportion of cytotoxic T lymphocytes (CTL) in the intestine and weakening the anti-tumor immunity of CAC. (D) necrosis and pyroptosis escalate when autophagy is deficient. N-GSDMD, RIPK3 (receptor interacting serine/threonine kinase 3), and MLKL (mixed lineage kinase domain like pseudokinase) can activate the MAPK/JNK pathway, contributing to the malignant transformation in CAC.
Table 1. Examples of autophagy modulators that inhibit colitis-associated colon cancer (CAC) development by promoting autophagy.