Figures & data
Table 1. Lipids involved in regulated cell death.
Figure 1. Lipid mechanism in apoptosis. Under intrinsic lethal stimuli, activated BAX or BAK1 oligomerizes and forms pores in the mitochondrial membrane, resulting in mitochondrial outer membrane permeabilization (MOMP) and the subsequent release of CYCS/cytochrome c into the cytosol. During this process, cardiolipin (CL) translocates to the mitochondrial outer membrane (MOM) and acts as a binding site for truncated BID (tBID), BAX or BAK1, triggering pore formation. Furthermore, mitochondrial ceramide (Cer) is necessary for BAX oligomerization. Cer and activated BAX together contribute to the formation of stable pores known as the ceramide channel (Cer-BAX channel). Additionally, sphingosine-1-phosphate (S1P) is degraded by SGPL1/S1P lyase to form hexadecenal. S1P and hexadecenal specifically cooperate with BAK1 and BAX, respectively. Conversely, cholesterol (Chol) accumulation in the mitochondrial membrane inhibits MOMP. Under extrinsic lethal stimuli, activated CASP8 can induce apoptosis directly by activating CASP3 or CASP7 or indirectly through the activation of the BH3-only protein BID, leading to the intrinsic apoptotic pathway. Lipid raft-disrupting agents such as methyl-beta cyclodextrin (MβCD), resveratrol, and edelfosine can trigger apoptosis. Cer, a hydrolyzed product of sphingomyelin (SM), induces coalescence of elementary rafts and enhances DISC formation, thereby amplifying FAS signaling. Furthermore, palmitoylation of TNFRSF10A/DR4 promotes TNFSF10/TRAIL-mediated apoptotic signaling. Phosphatidylserine (PS) exposure serves as an “eat me” signal for apoptotic cell clearance. [change TRAIL to “TNFSF10”.
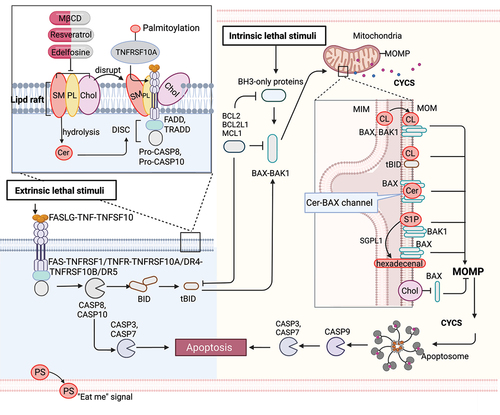
Figure 2. Lipid mechanism in necoroptosis. In the presence of caspase inhibition, necroptosis can be triggered by death receptor stimuli (e.g., TNFRSF1A/TNFR1 and FAS) or pathogen recognition receptor stimuli (e.g., TLR3 and TLR4). When CASP8 is inactive, a complex called the necrosome, comprised of RIPK1, RIPK3, and MLKL, forms. Within this complex, RIPK3-mediated phosphorylation induces a conformational change that activates MLKL, leading to its oligomerization and translocation to the plasma membrane. Subsequently, MLKL forms pores or cation channels at the plasma membrane, resulting in necroptosis. MLKL binds to PtdIns5P, PtdIns(4,5)P2, and PtdIns(3,4,5)P3, but not to unphosphorylated phosphatidylinositol (PtdIns) or other phospholipids. PIPs can be enzymatically deconstructed into diacylglycerol and inositol phosphates. MLKL interacts with highly phosphorylated inositol products (e.g., IP6, IP5, IP4) generated by IP kinases (IPMK and ITPK1). Necroptotic cells also expose phosphatidylserine (PS) on the outer leaflet of the plasma membrane, acting as an “eat-me” signal that can be recognized and phagocytosed.
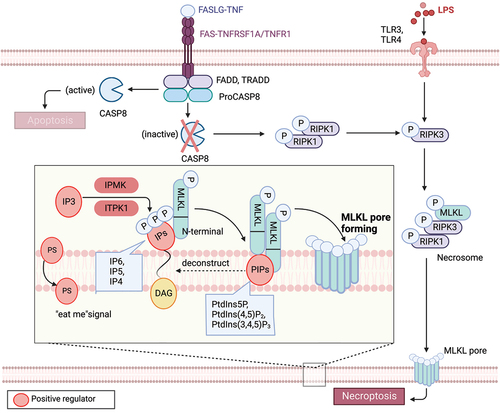
Figure 3. Lipid mechanism in ferroptosis. Ferroptosis is regulated by antioxidant enzymes, including GPX4 and AIFM2, which play a role in reducing ubiquinone (coenzyme Q10) and inhibiting ferroptosis. Among lipids, polyunsaturated fatty acids (PUFAs) are highly susceptible to peroxidation during ferroptosis. Long-chain saturated fatty acids (SFAs), such as palmitic acid (PA, C16:0) and stearic acid (SA, C18:0), as well as their downstream fatty alcohols (1-hexadecanol and 1-octadecanol), sensitize RSL3-induced ferroptosis. Conversely, exogenous monounsaturated fatty acids (MUFAs), including oleic acid (OA, C18:1) and palmitoleic acid (POA, C16:1), suppress ferroptosis. The regulation of key lipid metabolic enzymes involved in lipid synthesis, degradation, storage, transformation, and utilization is crucial in the context of ferroptosis. AA: arachidonic acid, AdA: adrenic acid, DHA: docosahexaenoic acid, DPA: docosapolyenoic acid, 1-HE: 1-hexadecanol, 1-OE: 1-octadecanol, eLPE/LPC: ether lysophosphatidylethanolamine/lysophosphocholine, ePe/epc: ether phosphatidylethanolamine/phosphocholine.
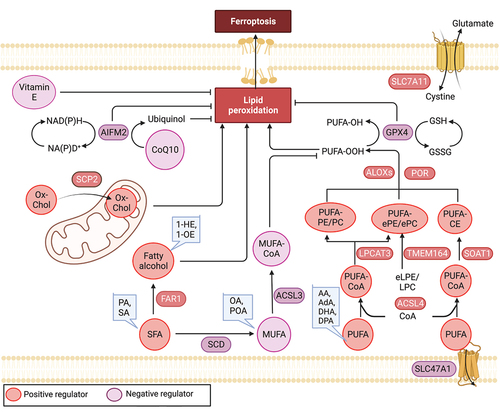
Figure 4. Lipid mechanism in pyroptosis. Pyroptosis is a proinflammatory form of programmed cell death characterized by the formation of membrane pores. The process of pore formation is executed through the inflammasome-mediated cleavage and activation of GSDMD. Lipids, including oxysterols, fatty acids, and phospholipids, play complex and critical roles in pyroptosis. For instance, the activation of the NLRP3 inflammasome requires binding to cholesterol in the endoplasmic reticulum, while the stable oligomerization of NLRP3 is facilitated by phosphoinositides (PIPs), and cardiolipin promotes NLRP3 inflammasome formation. Moreover, the functionality of GSDMD pores is dependent on phosphoinositides, and the enrichment of phosphatidylinositol-4,5-bisphosphate (PtdIns [Citation4,Citation5]P2) directly enhances their dynamics.
![Figure 4. Lipid mechanism in pyroptosis. Pyroptosis is a proinflammatory form of programmed cell death characterized by the formation of membrane pores. The process of pore formation is executed through the inflammasome-mediated cleavage and activation of GSDMD. Lipids, including oxysterols, fatty acids, and phospholipids, play complex and critical roles in pyroptosis. For instance, the activation of the NLRP3 inflammasome requires binding to cholesterol in the endoplasmic reticulum, while the stable oligomerization of NLRP3 is facilitated by phosphoinositides (PIPs), and cardiolipin promotes NLRP3 inflammasome formation. Moreover, the functionality of GSDMD pores is dependent on phosphoinositides, and the enrichment of phosphatidylinositol-4,5-bisphosphate (PtdIns [Citation4,Citation5]P2) directly enhances their dynamics.](/cms/asset/9e04df7f-e820-4ebe-ac24-e53b1a8b69f1/kaup_a_2259732_f0004_c.jpg)
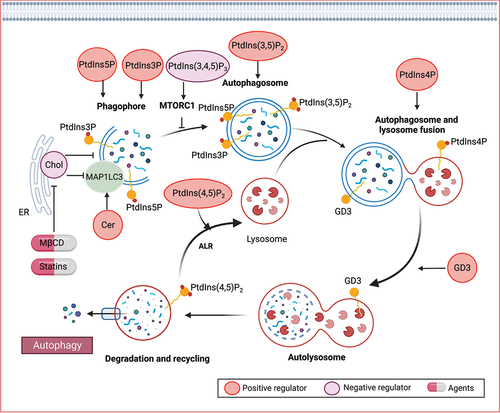
Figure 5. Lipid mechanisms in autophagy. Various lipid species, such as phospholipids, sphingolipids, and sterols, play crucial roles in different stages of autophagy. Specifically, phosphoinositides have been identified as key modulators of autophagy, involved in signaling processes that govern autophagy initiation, autophagosome biogenesis, and maturation. Ceramide (Cer) has also been implicated in the induction of autophagy. Furthermore, gangliosides, including GD3 gangliosides, can be recruited to autophagosomes and contribute to morphogenic remodeling.