
Figures & data
Figure 1. AMPK-deficient cells display increased apoptotic cell death with intact autophagy during prolonged amino acid deprivation. (A) Immortalized wild-type (WT) and prkaa1/AMPKα1 and prkaa2/AMPKα2 DKO MEFs (hereafter referred to as prkaa/AMPKα DKO MEFs) isolated from genetically modified mice were fed with complete media (+) for 1 h or amino acid-free media (-) for 0.5–16 h. Attached and floating cells were collected, and whole cell lysates were immunoblotted as indicated. ns, non-specific band detected by the PRKAA/AMPKα antibody. (B) Control and PRKAA1/AMPKα1 and PRKAA2/AMPKα2 (hereafter referred to as PRKAA/AMPKα) DKO HeLa cells generated by CRISPR-Cas9-mediated genome editing were treated as in (A). (C) MEFs were treated and amino acid deprived (16 h) as in (A), except ReadyProbe cell viability reagents were added to the culture media. Green fluorescence marks dead cells while blue (Dapi) fluorescence marks total cells. (D) Percent (%) cell death is indicated by the ratio of green cells:total blue cells. n = 3 fields of cells, each field containing ~700-900 cells from 1 representative experiment. Bars on graphs represent mean ± SD (standard deviation). Two-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; ***p < 0.001; ns, not significant. (E) WT and prkaa/AMPKα DKO MEFs were fed with complete media (+) for 1 h or amino acid-free media (-) for 1, 2, or 4 h in the absence or presence of BafA1. Whole cell lysates were immunoblotted as indicated. LE: light exposure; DE: dark exposure. ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (F) Control and PRKAA/AMPKα DKO HeLa cells were treated as in (E). (G) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa1/AMPKα1 and Prkaa2/AMPKα2 (hereafter referred to as Prkaa/AMPKα) siRNA and treated as in (E).
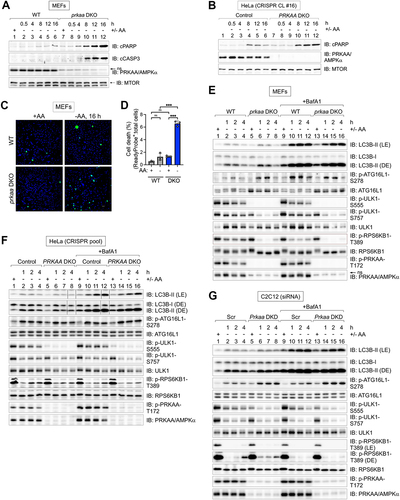
Figure 2. AMPK-deficient cells display increased autophagy induced by amino acid withdrawal. (A) WT and prkaa/AMPKα DKO MEFs were fed with complete (+) or amino acid-free media (-) for 2 h in the absence or presence of BafA1. ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (B) Quantification of LC3B-II:TUBA/α-Tubulin, LC3B-II:LC3B-I, and ATG16L1 p-S278:ATG16L1 from (A). n = 3 independent experiments. (C) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa/AMPKα siRNA and treated as in (A). (D) Quantification of LC3B-II:TUBA/α-Tubulin, LC3B-II:LC3B-I, and ATG16L1 p-S278:ATG16L1 from (C). n = 4 independent experiments. (E) WT and prkaa/AMPKα DKO MEFs fed with complete (+) or amino acid-free media (-) for 1–4 h in the absence of BafA1. The rate of degradation of SQSTM1/p62 was monitored by immunoblotting for SQSTM1/p62. (F) Quantification of SQSTM1:TUBA/α-Tubulin from (E). Normalized levels were set at 1.0 for wild-type and prkaa/AMPKα DKO MEFs. n = 3 independent experiments. (G) C2C12 cells were transiently transfected with siRNA as in (C) and treated as in (E) without BafA1. (H) Quantification of SQSTM1:TUBA/α-Tubulin from (G). Normalized levels were set at 1.0 for control and Prkaa/AMPKα DKD C2C12 cells. n = 3 independent experiments. (I) Model: AMPK suppresses (blue line) rather than promotes (red line) autophagy upon amino acid deprivation. In complete media, MTORC1 suppresses the initiation of autophagy. The rapid inhibition of MTORC1 by acute withdrawal of amino acids or treatment with torin1 initiates autophagy. Results in Fig. 1 and 2 indicate that AMPK is not required for autophagy induced by amino acid deprivation. In fact, AMPK may suppress autophagy in this context. Data shown as mean ± SD. Two-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
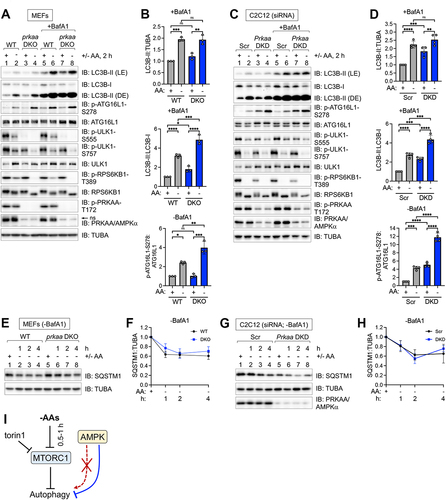
Figure 3. Autophagy induced by pharmacological MTORC1 inhibition occurs unimpaired in cells lacking AMPK. (A) WT and prkaa/AMPKα DKO MEFs were fed with complete media for 1 h and then treated without (-) or with (+) torin1 for 1–4 h in the absence or presence of BafA1. Whole cell lysates were immunoblotted as indicated. LE: light exposure; DE: dark exposure; ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (B) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa/AMPKα siRNA and treated as in (A). (C) Quantification of LC3B-II:TUBA/α-Tubulin, LC3B-II:LC3B-I, and ATG16L1 p-S278:ATG16L1 from (A) (2 h time point). n = 4 independent experiments. (D) Quantification of LC3B-II:TUBA/α-Tubulin, LC3B-II:LC3B-I, and ATG16L1 p-S278:ATG16L1 from (B) (2 h time point). n = 4 independent experiments. (E) WT and prkaa/AMPKα DKO MEFs were treated as in (A) without BafA1, and levels of SQSTM1/p62 were measured by immunoblotting. (F) Quantification of SQSTM1:TUBA/α-Tubulin from (E). Normalized levels were set at 1.0 for wild-type and prkaa/AMPKα DKO MEFs. n = 3 independent experiments. (G) C2C12 cells were transiently transfected with siRNA as in (B), treated as in (A) without BafA1, and analyzed as (E). (H) Quantification of SQSTM1:TUBA/α-Tubulin from (G). Normalized levels were set at 1.0 for control and Prkaa/AMPKα DKD C2C12 cells. n = 3 independent experiments. Data shown as mean ± SD. Two-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
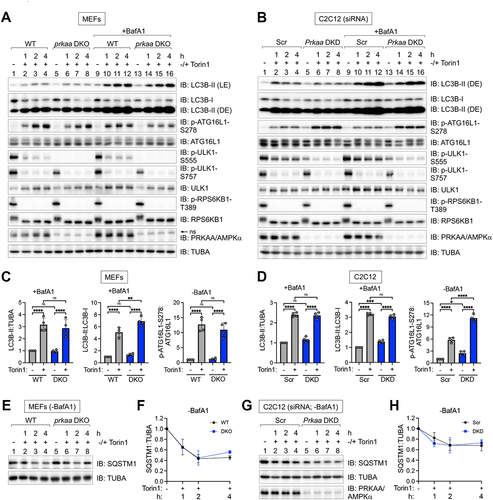
Figure 4. Induction of LC3B-, ATG16L1 p-S278-, and SQSTM1/p62-positive puncta by amino acid deprivation occurs unimpaired in AMPK-deficient cells, with evidence of increased autophagy in the absence of AMPK. (A) WT and prkaa/AMPKα DKO MEFs were fed with complete (+) or amino acid-free media (-) for 2 h in the presence of BafA1. Cells were fixed and stained for endogenous LC3B. Scale bar: 10 µm. (B) Quantification of LC3B puncta number per cell from (A). n = 30–40 cells from one experiment, representative of 4 independent experiments. (C) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa/AMPKα siRNA and treated as in (A). Scale bar: 10 µm. (D) Quantification of LC3B puncta number per cell from (C). n = 60–70 cells from two of four independent experiments. Below: Level of siRNA-mediated Prkaa/AMPKα knockdown in (C). (E) WT and prkaa/AMPKα DKO MEFs were fed with complete (+) or amino acid-free media (-) for 2 h in the absence of BafA1. Cells were fixed and stained for endogenous ATG16L1 p-S278. Scale bar: 5 µm. (F) Quantification of ATG16L1 p-S278 puncta number per cell from (E). n = 40 cells from two independent experiments. (G) C2C12 cells were transiently transfected with siRNA as in (C), fixed, and stained for endogenous ATG16L1 p-S278. Scale bar: 5 µm. (H) Quantification of ATG16L1 p-S278 puncta number per cell from (G). n = 30–40 cells from one of two independent experiments. Below: Level of siRNA-mediated Prkaa/AMPKα knockdown in (G). (I) WT and prkaa/AMPKα DKO MEFs were fed with complete (+) or amino acid-free media (-) for 2 h in the presence of BafA1. Cells were fixed and stained for endogenous SQSTM1/p62. Scale bar: 10 µm. (J) Quantification of SQSTM1 puncta number per cell from (I). n = 60–70 cells from 2 independent experiments. Bars on graphs represent mean ± SD. Two-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; ***p < 0.001; ****p < 0.0001; ns, not significant.
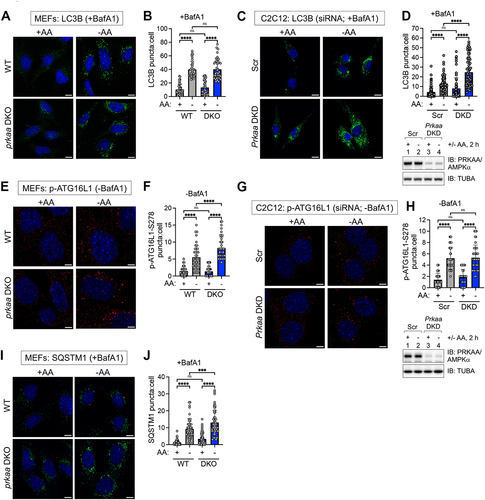
Figure 5. Formation of autophagosomal- and autolysosomal-like structures occurs unimpaired in AMPK-deficient cells upon amino acid deprivation. (A) WT and prkaa/AMPKα DKO MEFs were fed with complete (+AA) or amino acid-free media (−AA) for 2 h in the presence of BafA1. Cells were fixed, and the formation of autophagosomal- and autolysosomal-like structures was monitored by transmission electron microscopy (TEM). White arrowheads denote typical structures counted and quantified in (B). An enlarged view of the dotted box from the middle panel (−AA) is shown in the panels on the right (Enlarged view). Scale bar: 1 µm. (B) Quantification of the numbers of autophagosomal- and autolysosomal-like structures in (A). n = 40 fields of individual cells from one experiment. (C) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa/AMPKα siRNA and treated and analyzed as in (A). White arrowheads denote typical structures counted and quantified in (D). An enlarged view of the dotted box from the middle panel (−AA) is shown in the panels on the right (Enlarged view). Scale bar: 1 µm. (D) Quantification of the numbers of autophagosomal- and autolysosomal-like structures from (C). n = 40 fields of individual cells from one experiment. Below: Level of siRNA-mediated Prkaa/AMPKα knockdown in (C) was monitored by immunoblotting. Bars on graphs represent mean ± SD. Two-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; *p < 0.05; ****p < 0.0001; ns, not significant.
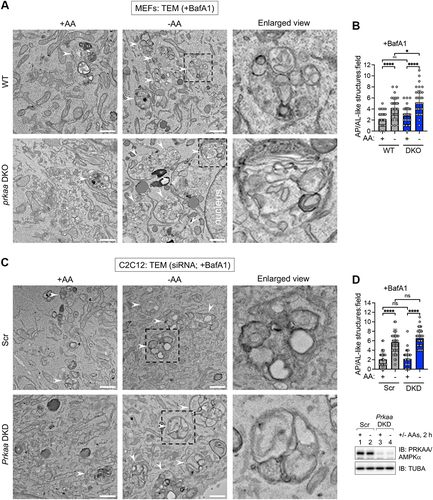
Figure 6. Activation of AMPK suppresses autophagy induced by amino acid deprivation. (A) WT and prkaa/AMPKα DKO MEFs were pre-treated without or with compound 991 (30 min) and fed with complete (+) or amino acid-free media (-) for 2 h in the absence or presence of 991 and the presence of BafA1. Whole cell lysates were immunoblotted as indicated. LE: light exposure; DE: dark exposure; ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (B) Quantification of LC3B-II:TUBA/α-Tubulin (upper graph) and LC3B-II:LC3B-I (lower graph) from (A). n = 4 independent experiments. (C) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa/AMPKα siRNA and treated as in (A). (D) Quantification of LC3B-II:TUBA/α-Tubulin (upper graph) and LC3B-II:LC3B-I (lower graph) from (C). n = 5 independent experiments. (E) WT MEFs were pre-treated without or with 991 (30 min) and fed with complete (+) or amino acid-free (-) media in the absence or presence of 991 and presence of BafA1 for 2 h. Cells were fixed and stained for endogenous LC3B. Scale bar: 10 µm. (F) Quantification of LC3B puncta number per cell from (E). n = 40 cells from 2 independent experiments. (G) WT C2C12 cells were treated as in (E). Scale bar: 10 µm. (H) Quantification of LC3B puncta number per cell from (G). n = 40 cells from 2 independent experiments. (I) WT and prkaa/AMPKα DKO MEFs were fed with complete (+) or amino acid-free (-) media for 2 h in the presence or absence of glucose (Glc) and the presence of BafA1. ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (J) Quantification of LC3B-II:TUBA/α-Tubulin (upper graph) and LC3B-II:LC3B-I (lower graph) from (I). n = 4 independent experiments. (K) C2C12 cells were transiently transfected with siRNA as in (C) and treated as in (I). (L) Quantification of LC3B-II:TUBA/α-Tubulin (upper graph) and LC3B-II:LC3B-I (lower graph l) from (K). n = 5 independent experiments. Bars on graphs represent mean ± SD. Two-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
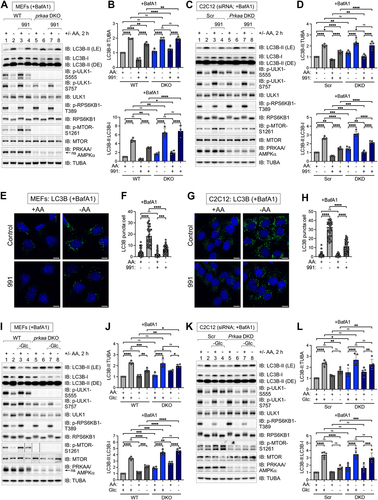
Figure 7. Activation of AMPK suppresses ULK1 signaling induced by amino acid deprivation while AMPK loss increases lysosomal acidification. (A) WT and prkaa/AMPKα DKO MEFs were pre-treated without or with compound 991 (30 min) and refed with complete (+) or amino acid-free (-) media for 2 h in the absence or presence of 991 and absence of BafA1. Whole cell lysates were immunoblotted as indicated. ns, non-specific band detected by the PRKAA/AMPKα antibody. (B) Quantification of ATG16L1 p-S278:ATG16L1 from (A). n = 4 independent experiments. (C) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa/AMPKα siRNA and treated as in (A). (D) Quantification of ATG16L1 p-S278:ATG16L1 from (C). n = 3 independent experiments. (E) WT and prkaa/AMPKα DKO MEFs were pre-treated without or with MRT68921 (30 min) and re-fed with complete (+) or amino acid-free (-) media for 2 h in the absence or presence of MRT68921 and absence of BafA1. (F) Quantification of ATG16L1 p-S278:ATG16L1 from (E). n = 4 independent experiments. (G) WT and prkaa/AMPKα DKO MEFs were fed with complete (+) or amino acid-free (-) media in the presence or absence of glucose (Glc) for 2 h and absence of BafA1. ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (H) Quantification of ATG16L1 p-S278:ATG16L1 from (G). n = 4 independent experiments. (I) C2C12 cells were transiently transfected with siRNA as in (C) and treated as in (G). (J) Quantification of ATG16L1 p-S278:ATG16L1 from (I). n = 3 independent experiments. (K) WT and prkaa/AMPKα DKO MEFs were fed with complete (+) or amino acid-free (-) media for 2 h in the absence of BafA1, treated with LysoTracker (30 min) and fixed. Scale bar: 10 µm. (L) Quantification of LysoTracker intensity per field (a.u., arbitrary unit) from (K). n = 10 fields of cells with 4–6 cells per field (~40–60 cells) from one experiment, representative of 2 independent experiments. (M) WT and Prkaa/AMPKα DKO C2C12 cells (CRISPR clone #16) were fed with complete (+) or amino acid-free (-) media for 2 h in the absence of BafA1, treated with LysoTracker (30 min) and fixed. Scale bar: 10 µm. (N) Quantification of LysoTracker intensity per field (a.u., arbitrary unit) from (M). n = 20 fields of cells with 15–20 cells per field (~300–400 cells) from one experiment, representative of 2 independent experiments. Bars on graphs represent mean ± SD. Two-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
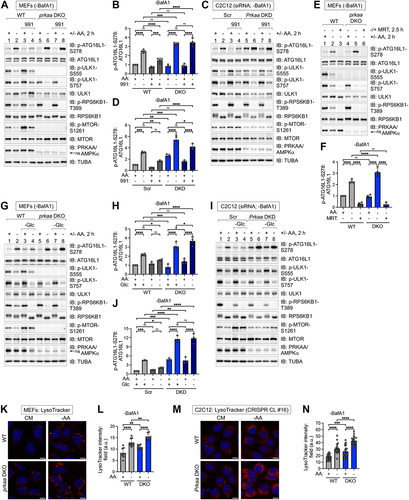
Figure 8. Activation of AMPK suppresses autophagic initiation and progression induced by pharmacological MTORC1 inhibition. (A) WT MEFs were pre-treated without or with rapamycin or torin1 (30 min), then pre-treated without or with 991 (30 min), and then re-fed with complete (+) or amino acid-free (-) media lacking or containing the drugs above for 2 h in the presence of BafA1. Whole cell lysates were immunoblotted as indicated (total treatment time, 3 h). LE, light exposure; DE, dark exposure. (B) Quantification of LC3B-II:TUBA/α-Tubulin (left panels) and ATG16L1 p-S278:ATG16L1 (right panels) from (A). Upper panels: torin1-treated cells; lower panels: rapamycin-treated cells. n = 4 independent experiments. (C) WT C2C12 cells were treated as in (A). (D) Quantification of LC3B-II:TUBA/α-Tubulin (left graphs) and ATG16L1 p-S278:ATG16L1 (right graphs) from (C). Upper panels: torin1-treated cells; lower panels: rapamycin-treated cells. n = 4 independent experiments. (E) WT and prkaa/AMPKα DKO MEFs were pre-treated without (-) or with (+) 991 (30 min) and then treated without (-) or with (+) torin1 in the absence or presence of 991 for 2 h and presence of BafA1. ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (F) Quantification of LC3B-II:TUBA/α-Tubulin and ATG16L1 p-S278:ATG16L1 from (E). n = 4 independent experiments. (G) C2C12 cells were transiently transfected with scrambled (Scr) or Prkaa/AMPKα siRNA and treated as in (E). (H) Quantification of LC3B-II:TUBA/α-Tubulin and ATG16L1 p-S278:ATG16L1 from (G). n = 4 independent experiments. (I) Model: Activation of AMPK with compound 991 suppresses autophagy induced by amino acid withdrawal, torin1, or rapamycin. Conversely, inactivation of MTORC1 with torin1 or rapamycin attenuates the ability of AMPK activation (using 991) to suppress autophagy. Bars on graphs represent mean ± SD. One-way ANOVA was performed followed by Sidak’s multiple comparison post hoc tests; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
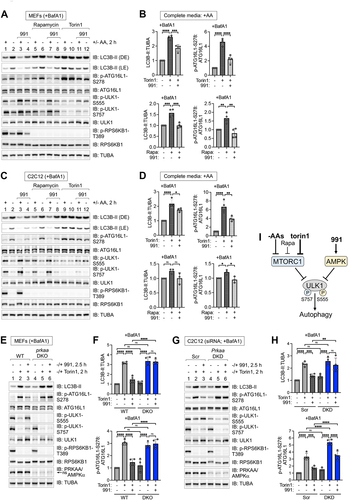
Figure 9. Impaired reactivation of MTORC1 signaling by the absence of AMPK during prolonged amino acid deprivation, and reduced ULK1 S555 phosphorylation upon amino acid withdrawal. (A) WT and prkaa/AMPKα DKO MEFs were fed with complete media (+) for 1 h or amino acid-free media (-) for 0.5–6 h. Whole cell lysates were immunoblotted as indicated. ns, non-specific band detected by the PRKAA/AMPKα antibody. (B) Control and PRKAA/AMPKα DKO HeLa cells were treated as in (A). (C) WT and prkaa/AMPKα DKO MEFs were fed with complete media (+) for 1 h or amino acid-free media (-) for 0.5, 2, or 4 h. Whole cell lysates were immunoblotted as indicated. ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (D) Quantification of RPS6KB1/S6K1 p-T389:RPS6KB1/S6K1 (left panels), EIF4EBP1/4EBP1 p-S65:EIF4EBP1/4EBP1 (middle panels), and ULK1 p-S757:ULK1 (right panels) from (C). n = 3 or 6 independent experiments. (E) Control and PRKAA/AMPKα DKO HeLa cells (CRISPR clone #16) were treated as in (C). (F) RPS6KB1/S6K1 p-T389:RPS6KB1/S6K1 (left panels), EIF4EBP1/4EBP1 p-S65:EIF4EBP1/4EBP1 (middle panels), and ULK1 p-S757:ULK1 (right panels) from (E). n = 4 or 6 independent experiments. (G) Quantification of ULK1 p-S555:total ULK1(upper panels) in MEFs or C2C12 cells cultured in complete media (+) or amino acid-free media (-) for 2 h. n = 5 independent experiments. Quantification of ULK1 p-S555:TUBA/α-Tubulin (lower panels) in MEFs or C2C12 cells cultured in complete media (+) or amino acid-free media (-) for 2 h. We also normalized ULK p-S555:TUBA/α-tubulin due to the extensive shifting that ULK1 undergoes on SDS-PAGE in response to amino acid levels (due to altered phosphorylation), rendering it difficult to assess total ULK1 levels accurately. n = 5 independent experiments. (H) WT and prkaa/AMPKα DKO MEFs were fed with complete media (CM) for 1 h or amino acid-free media (-) for 50 min and stimulated without (-) or with (+) amino acids for 10 min. ns, non-specific band detected by the PRKAA/AMPKα antibody in MEFs. (I) WT and prkaa/AMPKα DKO MEFs were amino acid-deprived (-) (50 min), pre-treated with torin1 (30 min), and stimulated without (-) or with (+) amino acids (10 min). (J) Summary: Both MTORC1 and AMPK suppress autophagy. In amino acid-replete media, MTORC1 phosphorylates ULK1 S757 while AMPK phosphorylates ULK1 S555 (and likely other sites) to suppress the activity of the ULK1 complex. AMPK also suppresses the acidification of lysosomes. The inactivation of MTORC1 by amino acid withdrawal or torin1 treatment reduces AMPK-mediated phosphorylation of ULK1. These events contribute to the activation of the ULK1 complex, which initiates the formation of autophagosomes. Upon induction of autophagy by amino acid deprivation, torin1, or rapamycin treatment, the activation of AMPK by compound 991, glucose withdrawal, or AICAR suppresses autophagy. Upon prolonged amino acid withdrawal, autophagic proteolysis raises levels of internal amino acids, which reactivate MTORC1 in a manner partly dependent on AMPK. Bars on graphs represent mean ± SD. Student’s t-tests (unpaired) were performed; *p < 0.05; **p < 0.01; ****p < 0.0001; ns, not significant.
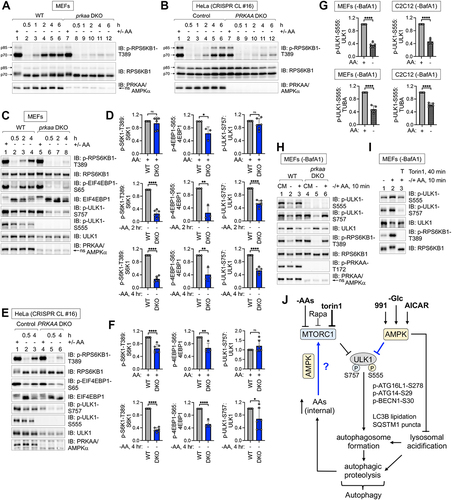
Figure 10. Models. (A) Revised model vs. prevailing view for how AMPK controls autophagy induced by amino acid withdrawal. (B) MTORC1- and AMPK-mediated phosphorylation of ULK1 during amino acid sufficiency stabilizes the AMPK-ULK1 interaction. In complete media, active MTORC1 phosphorylates ULK1 S757, stabilizing the AMPK-ULK1 interaction and facilitating the AMPK-mediated phosphorylation of ULK1 S555. These events inactivate the ULK1 complex. Upon amino acid withdrawal or torin1 treatment, the dephosphorylation of ULK1 S757 destabilizes the AMPK-ULK1 interaction, thus reducing the AMPK-mediated phosphorylation of ULK1 S555. These events activate the ULK1 complex. (C) Depending on the cellular context, AMPK exerts different effects on MTORC1 signaling. During energetic stress, AMPK inhibits MTORC1 signaling and thereby suppresses anabolic metabolism, which improves energy balance to promote cell survival. During nutrient stress caused by amino acid deprivation, AMPK contributes to the reactivation of MTORC1 signaling, which supports essential cellular processes to promote cell survival.
Supplemental materials v4_Kazyken et al R4.docx
Download MS Word (2.8 MB)Data availability statement
The data that support the findings of this study are contained in the manuscript and its Supplementary Materials files. The raw data are available from the corresponding authors upon reasonable request.