Figures & data
Figure 1. Genetic lesions involved in CLL clonal evolution and in Richter transformation. CLL is characterized by a high degree of molecular heterogeneity. During the clinical history of the disease, CLL may acquire new genetic lesions that may predispose to treatment resistance and, eventually, to Richter transformation. According to this model, at the time of diagnosis the CLL clone is characterized by a preponderance of cells that are sensible to chemotherapy (in yellow), and by only few cells that harbor genetic lesions, such as TP53 abnormalities, predisposing to chemo-refractoriness (in red). At the time of refractoriness to CIT, the CLL clone is mainly composed by cells (in red) that were not cleared by the previous chemotherapy because they harbor genetic lesions that confer refractoriness to CIT. Subsequent lines of treatment include BCRi (ibrutinib targeting BTK and idelalisib targeting PI3Kδ) and the BCL2i venetoclax. Despite the high efficacy of these drugs, a fraction of patients fails to respond over time. The mechanisms of refractoriness to ibrutinib and to venetoclax have been identified, at least to a certain extent. In particular, mutations of the BTK binding domain of ibrutinib, as exemplified in the cells colored in blue, and mutations of the BCL2 binding site of venetoclax, as exemplified in the cells colored in orange, may cause resistance to ibrutinib and to venetoclax, respectively. In addition, at every time point of the CLL clinical history, specific genetic abnormalities (in purple) involving the c-MYC, TP53, NOTCH1 and CDKN2A genes may predispose or lead to Richter transformation, represented by the histologic evolution of CLL to an aggressive B-cell lymphoma.
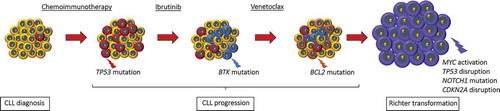
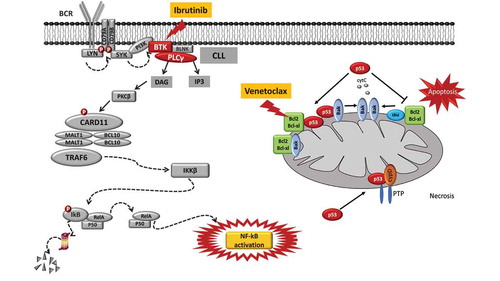
Figure 2. Molecular pathways targeted by ibrutinib and venetoclax and mechanisms of resistance to these drugs. Ibrutinib is a BTK inhibitor that acts by blocking the BTK catalytic site. The inactivation of the BCR pathway mediated by ibrutinib leads to the inhibition of the NF-κB pathway, thus reducing CLL cell proliferation and promoting apoptosis. The mode of action of ibrutinib is independent of TP53 disruption that represents the most robust predictor of chemo-refractoriness. Ibrutinib treatment may lead to the emergence of BTK missense mutations targeting codon 481 (p.C481S, leading to the cysteine-to-serine amino acid change) and thus altering the binding of the drug to BTK and causing the loss of its therapeutic effect. Venetoclax is a potent and selective BH3-mimetic drug that binds and blocks the BCL2 anti-apoptotic protein, leading to apoptosis that is independent of DNA damage response (and independent of disruption of TP53 that modulates the DNA damage response). A single nucleotide variant in the BCL2 gene (c.302G>T, p.Gly101Val) targets the BCL2 protein binding site of the drug, and may account for approximately 50% of cases of venetoclax resistance.