
Figures & data
Figure 1. TG-rich lipoprotein metabolism and novel drug targets. TG-rich lipoproteins are assembled from FA in the liver or intestine. CM are made in the small intestine and released into circulation via the lymphatic system, whereas VLDL is made in the liver and released directly into the blood stream. Both types of TG-rich lipoproteins are hydrolyzed by circulating LPL into respective remnant particles. CM remnants provide substrate for assembly of VLDL precursors in the liver, so completely impaired LPL function as in familial chylomicronemia syndrome is actually associated with deficiency of VLDL. In contrast, partially impaired LPL activity in multifactorial chylomicronemia syndrome is associated with elevations in CM, VLDL and their respective remnants. Several proteins modulate LPL activity and are targets for novel pharmacologic therapies for chylomicronemia. Most notably, apo C-III inhibits LPL activity and is the target of apo C-III antagonists (e.g. volanesorsen, olezarsen and ARO-APOC3). ANGPTL3 also inhibits LPL, as well as endothelial lipase activity, and antagonists of ANGPTL3 (e.g. evinacumab, vupanorsen and ARO-ANG3) are in development for the treatment of primary chylomicronemia. ANGPTL4 is another antagonist of LPL that may be a potential target for chylomicronemia treatment. Apo C-II is a co-factor for LPL and apo A-V stabilizes the LPL-apo C-II complex.
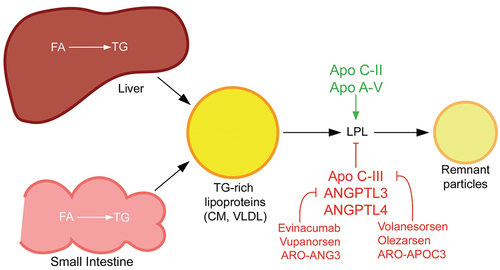
Table 1. Emerging treatments for chylomicronemia
Table 2. Discontinued treatments for chylomicronemia