
Figures & data
Figure 1. Population-based simulation framework for drug repurposing in the treatment of COVID-19.
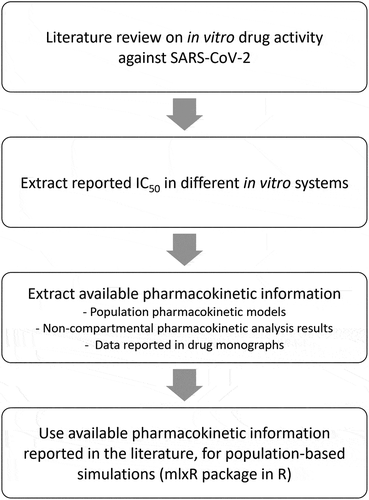
Table 1. Basic pharmacokinetic and pharmacodynamic parameters of each drug in different cell types.
Figure 2. Comparing the simulated plasma concentration–time profile of antimalarial drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of chloroquine were based on a published population pharmacokinetic model [Citation23] and compared with its IC50 in different cell lines [Citation17–19]. Chloroquine was assumed to be 60% bound to plasma proteins. (B) Simulated plasma concentrations of mefloquine were based on a population pharmacokinetic model and compared with its in vitro IC50 in different cell lines [Citation20,Citation29]. Mefloquine was assumed to be 98% bound to plasma proteins. Simulated plasma concentrations of amodiaquine (C) and its metabolite desethylamodiaquine (D) were based on a published population pharmacokinetic drug-metabolite model [Citation33] and compared with their in vitro IC50 in difference cell lines [Citation20,Citation29]. Both amodiaquine and desethylamodiaquine were assumed to be 90% bound to plasma proteins. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.
![Figure 2. Comparing the simulated plasma concentration–time profile of antimalarial drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of chloroquine were based on a published population pharmacokinetic model [Citation23] and compared with its IC50 in different cell lines [Citation17–19]. Chloroquine was assumed to be 60% bound to plasma proteins. (B) Simulated plasma concentrations of mefloquine were based on a population pharmacokinetic model and compared with its in vitro IC50 in different cell lines [Citation20,Citation29]. Mefloquine was assumed to be 98% bound to plasma proteins. Simulated plasma concentrations of amodiaquine (C) and its metabolite desethylamodiaquine (D) were based on a published population pharmacokinetic drug-metabolite model [Citation33] and compared with their in vitro IC50 in difference cell lines [Citation20,Citation29]. Both amodiaquine and desethylamodiaquine were assumed to be 90% bound to plasma proteins. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.](/cms/asset/cc6b85c9-3cbb-4b7d-98e7-1b146a9af08b/ierj_a_2113388_f0002_oc.jpg)
Figure 3. Comparing the simulated plasma concentration–time profiles of anti-hepatitis B drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of daclatasvir were based on a published population pharmacokinetic model of daclatasvir [Citation37] and compared with its IC50 in different cell lines [Citation18]. Daclatasvir was assumed to be 99% bound to plasma proteins. Simulated plasma concentrations of sofosbuvir (B) and its major metabolite, GS-331007 (C) were based on the population pharmacokinetic drug-metabolite model [Citation41] and compared with their in vitro IC50 in different cell lines [Citation18]. Sofosbuvir and GS-331007 was assumed to be 65% and 0% bound to plasma proteins, respectively. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.
![Figure 3. Comparing the simulated plasma concentration–time profiles of anti-hepatitis B drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of daclatasvir were based on a published population pharmacokinetic model of daclatasvir [Citation37] and compared with its IC50 in different cell lines [Citation18]. Daclatasvir was assumed to be 99% bound to plasma proteins. Simulated plasma concentrations of sofosbuvir (B) and its major metabolite, GS-331007 (C) were based on the population pharmacokinetic drug-metabolite model [Citation41] and compared with their in vitro IC50 in different cell lines [Citation18]. Sofosbuvir and GS-331007 was assumed to be 65% and 0% bound to plasma proteins, respectively. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.](/cms/asset/ece69970-8a54-4c4d-9620-fa56b4425049/ierj_a_2113388_f0003_oc.jpg)
Figure 4. Comparing the simulated plasma concentration–time profile of favipiravir (n = 1000) with its in vitro IC50 against SARS-CoV-2. Simulated plasma concentrations of favipiravir were based on a published population pharmacokinetic model of favipiravir [Citation46] and compared with its IC50 in Vero E6 cell line [Citation17]. Favipiravir was assumed to be 54% bound to plasma proteins. Solid black line represents the mean population plasma concentration–time profile, the shaded area represents the 90% prediction interval of the simulated concentrations, the blue line represents the uncorrected in vitro IC50 value, and the red line represents the in vitro IC50 value corrected for plasma protein binding.
![Figure 4. Comparing the simulated plasma concentration–time profile of favipiravir (n = 1000) with its in vitro IC50 against SARS-CoV-2. Simulated plasma concentrations of favipiravir were based on a published population pharmacokinetic model of favipiravir [Citation46] and compared with its IC50 in Vero E6 cell line [Citation17]. Favipiravir was assumed to be 54% bound to plasma proteins. Solid black line represents the mean population plasma concentration–time profile, the shaded area represents the 90% prediction interval of the simulated concentrations, the blue line represents the uncorrected in vitro IC50 value, and the red line represents the in vitro IC50 value corrected for plasma protein binding.](/cms/asset/cd50f2fc-cda4-4d44-b9b9-970678945f07/ierj_a_2113388_f0004_oc.jpg)
Figure 5. Comparing the simulated plasma concentration–time profiles of antiparasitic drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of tizoxanide were based on a published PBPK model of nitazoxanide [Citation52] and compared with its IC50 value reported in Vero E6 cells [Citation17]. Tizoxanide was assumed to be 99% bound to plasma proteins. (B) Simulated plasma concentrations of ivermectin were based on a published population pharmacokinetic model of ivermectin in healthy volunteers [Citation60] and compared with its IC50 values reported in Vero-hSLAM cells [Citation62]. Ivermectin was assumed to be 93% bound to plasma proteins. (C) Simulated atazanavir plasma concentrations were based on a published population pharmacokinetic model of atazanavir in HIV infected patients [Citation68] and compared with its IC50 in different cell lines [Citation64]. Atazanavir was assumed to be 86% bound to plasma proteins. (D) Simulated colchicine plasma concentrations were based on subject-specific nonlinear regression model [Citation75] and compared with plasma concentrations inhibiting neutrophil chemotaxis [Citation79]. Solid black lines represent the mean population concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.
![Figure 5. Comparing the simulated plasma concentration–time profiles of antiparasitic drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of tizoxanide were based on a published PBPK model of nitazoxanide [Citation52] and compared with its IC50 value reported in Vero E6 cells [Citation17]. Tizoxanide was assumed to be 99% bound to plasma proteins. (B) Simulated plasma concentrations of ivermectin were based on a published population pharmacokinetic model of ivermectin in healthy volunteers [Citation60] and compared with its IC50 values reported in Vero-hSLAM cells [Citation62]. Ivermectin was assumed to be 93% bound to plasma proteins. (C) Simulated atazanavir plasma concentrations were based on a published population pharmacokinetic model of atazanavir in HIV infected patients [Citation68] and compared with its IC50 in different cell lines [Citation64]. Atazanavir was assumed to be 86% bound to plasma proteins. (D) Simulated colchicine plasma concentrations were based on subject-specific nonlinear regression model [Citation75] and compared with plasma concentrations inhibiting neutrophil chemotaxis [Citation79]. Solid black lines represent the mean population concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.](/cms/asset/3b4f5ff2-eb17-4df9-aa92-d001531afd1e/ierj_a_2113388_f0005_oc.jpg)