Figures & data
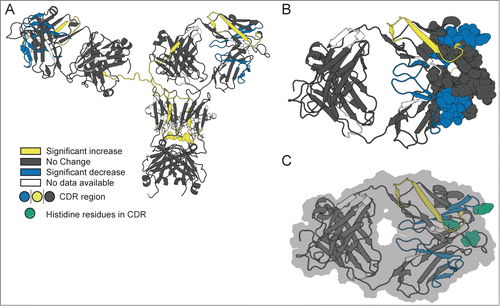
Figure 1. Hydrodynamic diameter of mAb-C under various solution conditions as measured by dynamic light scattering. (A) Effect of pH as a function of protein concentration. (B) Effect of salt type (NaCl and Na2SO4 and salt concentration. Experiments in panel A were conducted at 25°C with mAb-C samples prepared in 40 mM potassium phosphate buffer (pH 6, 7 and 8) containing 0.3 M NaCl. Experiments in panel B were conducted at 25°C with mAb-C samples prepared in 40 mM potassium phosphate buffer (pH 7) containing either NaCl or Na2SO4 at 0, 0.15, 0.3 and 0.5 M concentration. The error bars represent one standard deviation from 3 independent measurements.
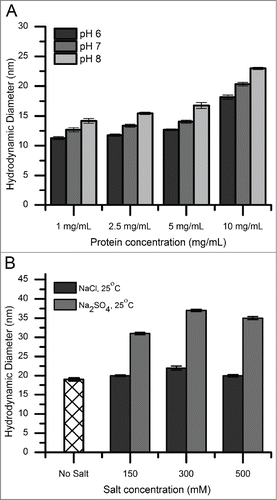
Figure 2. Cross-linking and SDS-PAGE analysis of reversible self-association (RSA) of mAb-C under different solution conditions. The mAb-C samples were prepared in 40 mM potassium phosphate buffer (pH 7) in the presence and absence of 0.3 M Na2SO4. The lane in each panel marked “UN” represents a mAb-C control with no added cross-linker. The first and last lane of each gel contains molecular weight standards. The masses are denoted on the left side gels. Subsequent lanes show the extent of mAb-C cross-linking in the presence of increasing molar ratio of BS2G cross-linker. Top-left panel, non-reduced SDS-PAGE gel showing cross-linking of mAb-C in the absence of Na2SO4. Top-right panel, non-reduced gel showing cross-linking of mAb-C in the presence of 300 mM Na2SO4. Bottom-left panel, reduced SDS-PAGE gel showing mAb-C in the absence of Na2SO4. Bottom-right panel, reduced gel of mAb-C in the presence of 300 mM Na2SO4. Cross-linking reactions were carried out at 4°C.
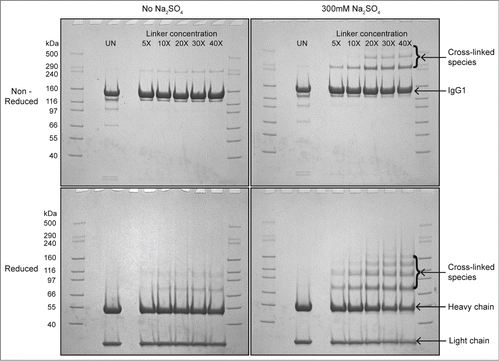
Figure 3. The effect of protein concentration, temperature and salt type on solution viscosity of mAb-C samples either before or after lyophilization and reconstitution. (A) Effect of temperature and salt type on viscosity of mAb-C as a function of protein concentration. (B) Effect of lyophilization and reconstitution diluent (with H2O- and D2O-based buffers) on the viscosity of mAb-C as a function of protein concentration. Samples of mAb-C for panel A were prepared in 40 mM potassium phosphate buffer (pH 7.0) containing 300 mM salt (NaCl or Na2SO4). For panel B, mAb-C samples that were not lyophilized were prepared in 40 mM potassium phosphate buffer (pH 7.0) containing 300 mM Na2SO4 and 10% (w/v) trehalose. Lyophilized samples were freeze-dried in 20 mM potassium phosphate buffer (pH 7.0) containing 10% (w/v) trehalose, and then reconstituted with either H2O or D2O buffers consisting of 20 mM potassium phosphate buffer (pH 7.0) and 300 mM salt Na2SO4. Viscosity measurements were taken either at 4°C or 25°C, as noted. The error bars represent one standard deviation from 3 independent measurements.
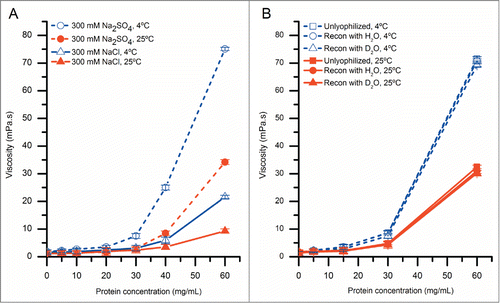
Figure 4. Circular dichroism spectra showing the effect of the lyophilization process on the overall secondary structure of mAb-C. All mAb-C samples (pre and post lyophilization) were diluted to 0.3 mg/mL with 40 mM potassium phosphate buffer (pH 7.0) containing 300 mM Na2SO4 and 10% (w/v) trehalose for analysis at 10°C. The error bars represent one standard deviation from 3 independent measurements.
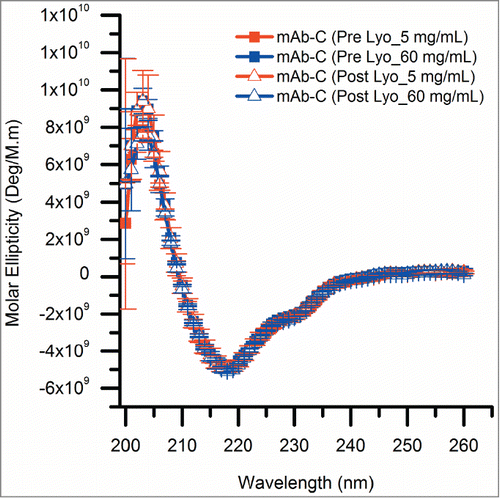
Table 1. Effect of lyophilization and reconstitution on the aggregation profile of mAb-C as measured by size-exclusion chromatography (SEC). Samples of mAb-C were prepared at 5 and 60 mg/mL in 20 mM potassium phosphate buffer (pH 7.0) with 10% (w/v) trehalose (pre-lyophilization samples). After lyophilization, mAb-C samples were reconstituted with D2O-based 20 mM potassium phosphate buffer (pH 7.0) with 300 mM Na2SO4 (post-lyophilization samples). The experimental data are mean and standard deviation calculated from three independent measurements on three separate lyophilized vials.
Figure 5. Deuterium uptake by 12 representative peptide segments from mAb-C measured at 5 and 60 mg/mL as determined by HX-MS. (A) Six representative peptides that showed no differences in hydrogen exchange kinetics between low and high protein concentrations. (B) Six representative peptides that showed significant changes in hydrogen exchange kinetics between low and high protein concentrations. Domain location and peptide number of the segment are shown in parentheses. The error bars represent one standard deviation from 3 independent experiments. Refer to Figure S2 for deuterium uptake plots of all 130 peptides in the data set.
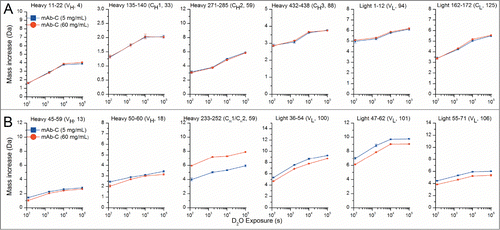
Figure 6. Relative differences in deuterium uptake at 4 exposure times as measured by HX-MS for 130 peptide segments of mAb-C at 60 mg/mL vs. 5 mg/mL at pH 7.0. The individual peptides are arranged on the horizontal axis starting from the N-terminal of the heavy chain and ending at the C-terminal of the light chain. The peptides are numbered sequentially based on the locations of their middle residues (see Table S1 for the identities and locations of the peptides). The horizontal axes of these plots denote the peptide numbers from 1 to 130. The vertical axis is the difference between exchange at 60 mg/mL vs. 5 mg/ml: . Positive bars indicate an increase in deuterium uptake for a particular peptide segment at high protein concentration (60 mg/mL) and negative bars indicate decreased deuterium uptake for a peptide segment at 60 mg/mL compared to lower protein concentration (5 mg/mL). The dashed lines at ± 0.4 Da indicate the 99% confidence limits for significant differences. White and gray shades in the background of the figure represent IgG domain boundaries and each domain is labeled at the top of the figure. Shades in blue represent CDR segments on mAb-C. Segment locations in the mAb-C sequence and their corresponding peptide numbers can be found in Table S1 in the supporting information. An average of 3 independent mass measurements was used to calculate each mass difference data point corresponding to all the exposure time-points.
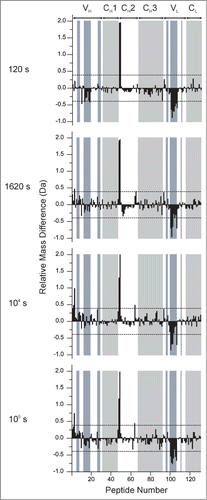
Figure 7. Effect of concentration-dependent RSA on deuterium uptake of various segments of mAb-C as measured by HX-MS plotted onto a homology model of mAb-C. (A) Entire mAb, (B) view of the Fab domain and (C) view of the Fab domain with histidine residues in the peptide segments containing the CDR2H and CDR3H sequences highlighted in green. Changes in deuterium uptake of particular peptide segments are colored according to the legend and are derived from the differential exchange data shown in .