Figures & data
Figure 1. Comparison of regions that change in DNA accessibility upon stimulation to those that do not. (A) The MCF-7 model of EMT and CSC formation. Non-stimulated (NS) cells are treated with PMA (ST) to produce mesenchymal cells with non-CSC (NCSC) and CSC properties. (B) Accessible regions were grouped according to accessible in NS and ST cells, only accessible in NS or ST (only), more accessible in NS or ST (more), or equal accessibility. Regions for each group were annotated by distance to the nearest transcription start site (TSS) (B), genomic region (C), summarised chromatin environment in different cell types and (D) summarised CTCF/chromatin in MCF-7 and mammary epithelial cells (HMEC) (E). For summarised chromatin (D, E), each region is colored by their state. DNA accessibility at specific regions (F). Dots show biological replicates, dashes show averages. FAIRE-PCR normalized to PPIA and scaled to average of NS. purple: ST, orange: MDA-MB-231, * p < 0.05 compared to NS.
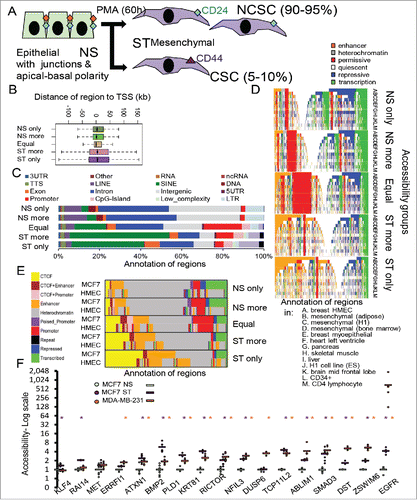
Figure 2. Identification of the regions with increased accessibility that have changes in enhancer histone marks and are within certain distances of genes with differential expression. (A) Enhancer histone marks around the regions with different accessibility ordered by strength of accessibility. Enhancer marks are shown for non-stimulated and PMA-stimulated (ST) MCF-7 cells and NS MDA-MB-231 (MDA) cells. (B) For the ST only accessibility regions, marks were clustered into different patterns and the chromatin state of the regions in MCF-7 cells annotated. The average ChIP and FAIRE-seq profiles are shown for each subset of the resulting clusters with the number of regions in each cluster indicated. (C) Genes nearest to the regions were profiled for enrichment of Gene Ontology, Reactome, and KEGG pathways. The significance of the enrichment of different gene groups is indicated. (D) Number of regions with accessibility only in stimulated MCF-7 cells within different distances of genes with differential expression in NS and ST cells or non-cancer stem cells (NCSCs) and CSCs. Error bars show SD of 100 sets of random genes with the sets the same size as the gene set of interest. * z-score > 2. Marks are shown +/− 1 kb around the region, reads binned by 0.1 kb and adjusted by total mapped reads (A,B).
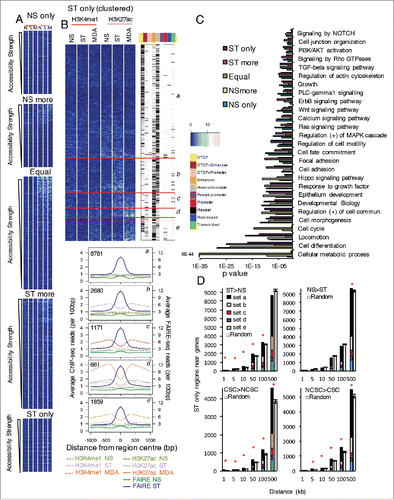
Figure 3. Identification of the DNA binding motifs in regions with increased accessibility and how the motifs occur in the regions with respect to expression of nearby genes. (A) The proportion of regions with differential or equal (Eq.) accessibility containing motifs for AP1 (FOSL1), FOX (FOXA2), CTCF, TFAP (AP-2G), and TEAD (TEAD4) compared to background regions. (B) Expression in NS, NCSCs, and CSCs of the factors that can bind the motifs. (C) The transcription factor motifs enriched in regions near to genes with differential expression and with accessibility only in ST MCF-7 cells. Expression was compared between (i) NS and ST and (ii) NCSCs and CSCs. CLOVER enrichment scores are shown in brackets after the motif colored by the comparison they are significant in. All motifs with a score had p-value< 0.05 for enrichment when the regions within 50 kb of a gene in the gene set were compared to all regions with ‘ST only’ accessibility (C). * q-value (Benjamini) = 0 and p < 10−15 (A).
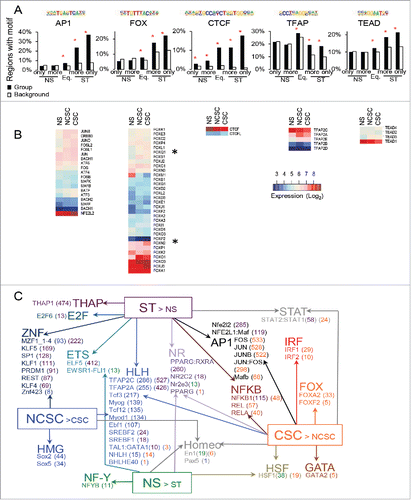
Figure 4. Expression and localization of FOXN2 and FOXQ1 and their role in regulating the CSC phenotype. (A) Expression of FOXN2 and FOXQ1 in non-stimulated (NS), PMA-stimulated (ST), adherent (AD), and floating (FLT) cells. (B) Levels of FOXN2 and FOXQ1 in the nucleus as measured by total nuclear fluorescence intensity (TNFI) and the nuclear/cytoplasmic ratio (Fn/c) in the MCF-7 model and MDA-MB-231 (MDA) cells. (C) The effect of FOXN2 and FOXQ1 siRNA on the CD44Hi and CH24lo proportion of ST-AD and ST-FLT MCF-7 cells compared to control ST (Ctrl). (D) The effect of FOXN2 and FOXQ1 siRNA on the levels of laminin V and cytoplasmic fibronectin in MCF-7 cells. Expression normalized to PPIA, average of n = 3 +/− SEM (A). For protein localization, DAPI was used to identify the nucleus, image scale bar is 10 μm, and averages of 20 cells are shown +/− SEM * for p ≤ 0.05, ** for p ≤ 0.01, *** for p ≤ 0.001, and **** for p ≤ 0.0001. (B, D). FACS n = 3, error bars = SEM, red stars show significance against control siRNA (C).
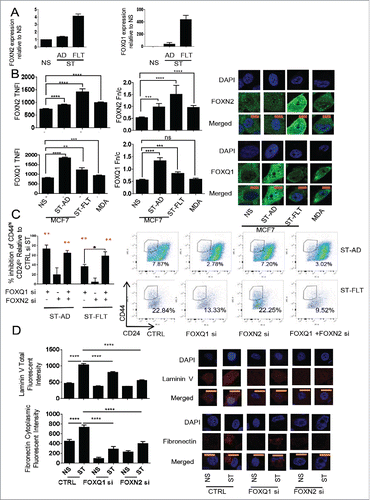
Figure 5. Putative FOX targets in cancer stem cell (CSC) gene expression. (A-D) Expression of genes with increased expression in CSCs than NCSCs that have regions with increased accessibility upon stimulation and that contain FOX binding motifs. Genes are grouped A-D by how H3K27ac differs in ST and MDA-MB-231s (see ). Regions could contain any of 3 variants of the core primary FOX motif. Microarray expression values are scaled (z-score). (E) The level of inhibition of expression due to FOXN2 and FOXQ1 siRNA for putative targets in MCF-7 cells stimulated (ST) with PMA. PCR expression normalized to PPIA and expressed as percent inhibition of the control siRNA ST value, n = 3 PCR replicates, error bars SEM (E).
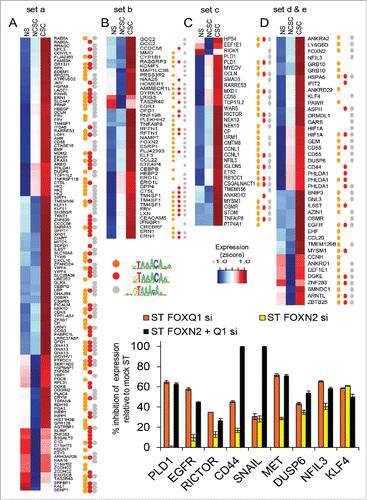
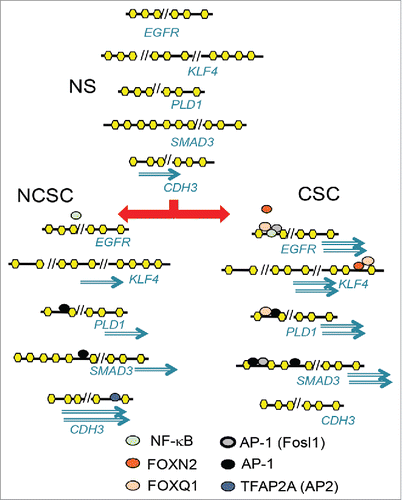
Figure 6. Putative models of how the factors binding the motifs may act differently in NS, NCSCs, and CSCs to give rise to genes with higher expression in CSCs than NCSCs or NCSCs than CSCs. Upon stimulation of MCF-7 cells (NS), transcription factors such as AP-1 increase chromatin accessibility at regulatory regions away from the transcription start site. The differential activation of AP-1 and FOX members and TFAP2A in cancer stem cells (CSC) and non-CSC (NCSC) regulates differential gene expression. Genes can have more than one regulatory region that become accessible to different extents in NCSC and CSC, giving rise to different levels of expression.