Figures & data
Table 1. Novel Pathogenic mechanisms adopted by P. gingivalis to exaggerate periodontal inflammation.
Figure 1. Virulence factors of P. gingivalis: P gingivalis has various virulence factors that help it to invade the host cell and evade the defense mechanisms of the host. Some of the most important adhesions of P. gingivalis include Lipopolysaccharide (LPS), Capsule, outer Membrane protein, Peptidoglycan, Major, and minor Fimbriae and Pilli. P. gingivalis also release enzymes and cytotoxic molecules such as defensins, autoinducer proteins (AI-2); 8 hydroxy 2ʹ deoxyguanosine, superoxide anions, matrix metalloproteinases, caspase-3, catalase, nucleotide-diphosphate-kinase. These enzymes help to invade the host cell, increase oxidative stress, and enhance biofilm formation. P. gingivalis is also known to release endogenous antioxidants such as thiol, peroxidase, glutathione, superoxide dismutase, rubrerythrin to protect itself from the surrounding free radicals.
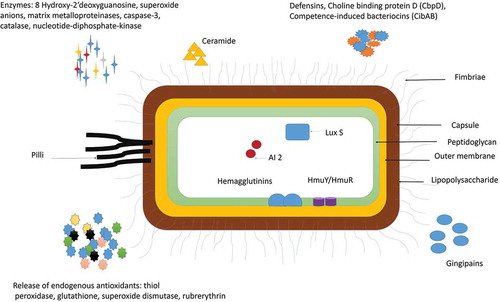
Figure 2. P. gingivalis modulates the host immune response by facilitating the growth of pathobionts species and altering the function of various immune cells of the host. The weakened immune response enhances biofilm formation and oxidative stress that in turn increases the periodontal inflammation and favors the growth of P. gingivalis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells]. [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- Interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells; NOD-nucleotide-binding oligomerization domain; NF-Kb – Nuclear Factor kappa Beta].
![Figure 2. P. gingivalis modulates the host immune response by facilitating the growth of pathobionts species and altering the function of various immune cells of the host. The weakened immune response enhances biofilm formation and oxidative stress that in turn increases the periodontal inflammation and favors the growth of P. gingivalis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells]. [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; ROS-Reactive oxygen species; MMPS- matrix metalloproteinase; IL- Interleukin; RANKL- Receptor activator of nuclear factor-kappa beta; TNF- Tumor Necrotic Factor-alpha; Th – T helper cells; NOD-nucleotide-binding oligomerization domain; NF-Kb – Nuclear Factor kappa Beta].](/cms/asset/e7174048-18ff-43d2-b58e-847c46a08305/zjom_a_1801090_f0002_oc.jpg)
Figure 3. Interaction of P. gingivalis with F. alocis and its effects on biofilm formation and periodontal inflammation: P. gingivalis interaction with F. alocis can modulate the innate immune response of the host as both these species auto-aggregate and express unique genes expression. P. gingivalis-F. alocis remodeling the actin and chromatin molecule, activation of autoinducer (AI) associated quorum sensing, the proliferation of the junctional epithelium, and deposition of collagen fibers in the gingival epithelial cells. The co-infection also upregulates the production of extracellular matrix adhesion proteins, CRISPRs RNA, and toxin-antitoxin system proteins that increase the adherence and colonization of P. gingivalis with other ‘Gram-positive bacteria and host tissues’, and are directly linked with increased biofilm formation. The CRISPR-RNA also helps with triggering the stress response, chaperone formation, and horizontal gene transfer among oral bacteria that favor microbial community development.
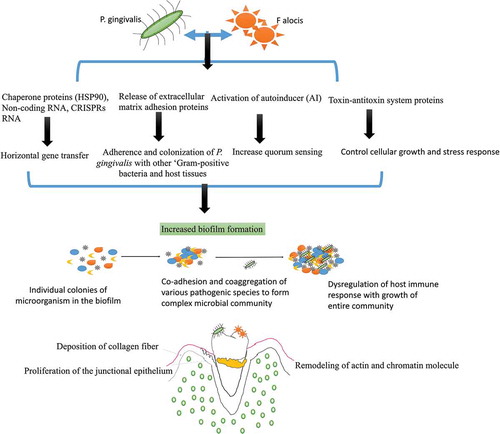
Figure 4. Modulation of immunoinflammatory response by two forms of P. gingivalis lipid A depending on the microenvironment (hemin level) and their interference in TLR4 receptor signaling downstream activation. It can regulate the inflammatory response according to changes in the environmental conditions by changing its LPS moiety [Abbreviation: LPS: lipopolysaccharide; p65: nuclear factor NF-κB protein p65 subunit; p50: nuclear factor NF-κB protein p50 subunit; TLR4: Toll-like receptor-4; TRAF 6: tumor necrosis factor receptor-associated factor 6; TRIF: TIR-domain-containing adapter-inducing interferon-β; TRAM: TRIF-related adaptor molecule].
![Figure 4. Modulation of immunoinflammatory response by two forms of P. gingivalis lipid A depending on the microenvironment (hemin level) and their interference in TLR4 receptor signaling downstream activation. It can regulate the inflammatory response according to changes in the environmental conditions by changing its LPS moiety [Abbreviation: LPS: lipopolysaccharide; p65: nuclear factor NF-κB protein p65 subunit; p50: nuclear factor NF-κB protein p50 subunit; TLR4: Toll-like receptor-4; TRAF 6: tumor necrosis factor receptor-associated factor 6; TRIF: TIR-domain-containing adapter-inducing interferon-β; TRAM: TRIF-related adaptor molecule].](/cms/asset/30926e1f-2e89-48fa-a0e4-bb4543026300/zjom_a_1801090_f0004_oc.jpg)
Figure 5. Schematic representation of the interaction between P. gingivalis and S. gordonii: P. gingivalis interacts with S. gordonii by utilizing its major (FimA) and minor fimbriae (Mfa1). FimA and Mfa1 of P. gingivalis binds to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and streptococcal SspA/B adhesins (SspA/B termed BAR antigen I/II) of S. gordonii, respectively The Mfa1 interactions with SspB lead to phenotypic changes with subsequent activation of tyrosine phosphorylation signal production (PTK). The increased PTK signaling causes exopolysaccharide formation that causes accretion of P. gingivalis colonies. The FimA also interacts to the α5β1-integrin receptors on gingival epithelial to facilitate bacterial entry.
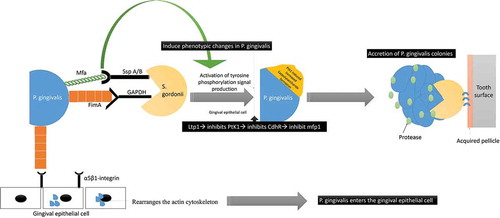
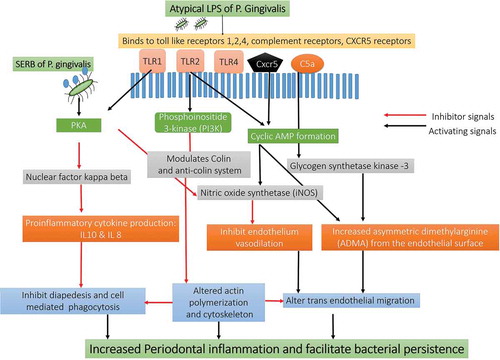
Figure 6. Schematic representation Immune response pathways triggered by the activation of TLR2/TLR4/CXCR5/C5aR receptors by gingipains P. gingivalis: The gingipains of P. gingivalis degrade the C5 and C3 from the complement system and degrade them C5a and C3a. The C5a interacts with the receptors on the neutrophils, epithelial cells, and endothelium in the host to Impairs phagocytosis and increase proinflammatory cytokine. C3a also inhibits the caspase 11–dependent non-canonical inflammasome pathway and prevents the apoptosis of the cell and allows P. gingivalis to use the host cell for its growth. The C5aR-TLR2 cross-talk activated by P. gingivalis pili induced degradation of MyD88 in neutrophils. In the absence of MyD88, the co-association of C5aR-TLR2 promotes P. gingivalis infection v activation of the TIRAP-dependent PI3 K signaling pathway. This, in turn, causes an inflammatory cytokine TNF-α along with inhibition of RhoA activation and actin polymerization that impairs the process of the maturation of phagosomes and P. gingivalis phagocytosis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; CXCR4: C-X-C chemokine receptor type 4; cAMP: cyclic adenosine monophosphate; iNOS: inducible nitric oxide synthase; Mal: MyD88 adapter-like; p38MAPK: mitogen-activated protein kinase p38; PKA: protein kinase A; PI3 K: phosphoinositide-3-kinase; RhoA: Ras homolog gene family, member A; sTREM-1 – sTREM-1 – salivary Triggering Receptor Expressed on Myeloid cells 1ʹ Protease-activated receptors [PAR]-2].
![Figure 6. Schematic representation Immune response pathways triggered by the activation of TLR2/TLR4/CXCR5/C5aR receptors by gingipains P. gingivalis: The gingipains of P. gingivalis degrade the C5 and C3 from the complement system and degrade them C5a and C3a. The C5a interacts with the receptors on the neutrophils, epithelial cells, and endothelium in the host to Impairs phagocytosis and increase proinflammatory cytokine. C3a also inhibits the caspase 11–dependent non-canonical inflammasome pathway and prevents the apoptosis of the cell and allows P. gingivalis to use the host cell for its growth. The C5aR-TLR2 cross-talk activated by P. gingivalis pili induced degradation of MyD88 in neutrophils. In the absence of MyD88, the co-association of C5aR-TLR2 promotes P. gingivalis infection v activation of the TIRAP-dependent PI3 K signaling pathway. This, in turn, causes an inflammatory cytokine TNF-α along with inhibition of RhoA activation and actin polymerization that impairs the process of the maturation of phagosomes and P. gingivalis phagocytosis [Abbreviation: CR- complement receptors; TLR- Toll-like receptors; CXCR4: C-X-C chemokine receptor type 4; cAMP: cyclic adenosine monophosphate; iNOS: inducible nitric oxide synthase; Mal: MyD88 adapter-like; p38MAPK: mitogen-activated protein kinase p38; PKA: protein kinase A; PI3 K: phosphoinositide-3-kinase; RhoA: Ras homolog gene family, member A; sTREM-1 – sTREM-1 – salivary Triggering Receptor Expressed on Myeloid cells 1ʹ Protease-activated receptors [PAR]-2].](/cms/asset/a45cf5e1-ceca-4546-b260-32a3f2c4ac3b/zjom_a_1801090_f0006_oc.jpg)
Figure 7. Schematic presentation of the immunological pathways triggered by LPS of P.gingivalis.