Figures & data
Figure 1. Strand-specific RNA-Seq reads aligned to the Schizosaccharomyces japonicus genome as viewed in the Broad's Integrative Genomics Viewer. Strand-specific RNA-Seq reads are shown aligning to the top strand (top) and bottom strand (center) separately. The left and right RNA-Seq paired fragment reads are colored red and light blue, respectively. The reference gene structure annotations for this 11-kb region of the genome is shown at bottom colored dark blue.
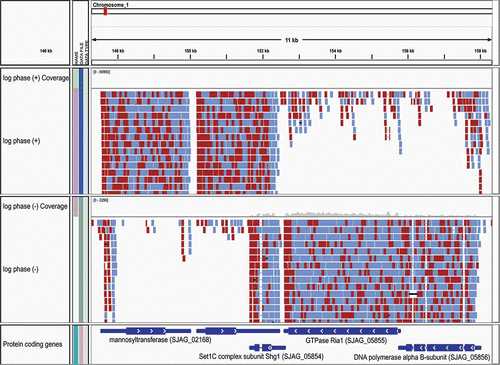
Figure 2. Hybrid approach to RNA-Seq-based transcript reconstruction leveraging genome alignment and de novo assembly. RNA-Seq reads are first aligned to the genome, then partitioned into disjoint regions of alignment coverage. Inchworm is leveraged to de novo assemble the read sequences into transcripts. The resulting transcripts are aligned to the genome using a conventional cDNA alignment tool, and PASA is leveraged to further assemble overlapping alignments and extract gene structure annotations.
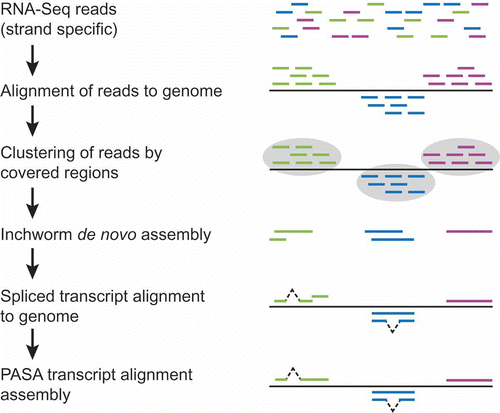
Figure 3. Spliced nucleotide and protein alignments infer intron structures. A section of AAT Alignments of homologous protein and EST sequences to the Neurospora crassa gene (shown as query) for alkaline phosphatase (NCU01376). This region of the alignment unambiguously identifies an intron within the gene structure; consensus splice sites are shown in bold.

Figure 4. ARGO genome annotation editor display. Shown is the evidence for the gene structure annotation of Neurospora crassa alkaline phosphatase (NCU01376) in the ARGO genome annotation editor. Evidence consists of, from top to bottom, Augustus, geneid, FgeneSH, SNAP, GLIMMERHMM, and GENEMARK.hmm ab initio predictions, followed by GENEWISE predictions based on top matching homologous proteins, PASA assemblies of EST alignments (ESTs not shown), EVidenceModeler consensus prediction, and the final annotated gene model for this locus. The intron boundaries that agree with the annotated gene model are highlighted as pink vertical bars. Positions of tart and stop codons are shown as green and red vertical bars, respectively. The ab initio predictors AUGUSTUS, FgeneSH, SNAP, and GENEMARK.hmm all perfectly agree on the structure of this gene, whereas GeneId and GLIMMERHMM propose different structures. The PASA assemblies of high quality EST alignments provide evidence for UTR annotations at both gene termini, extending upstream and downstream of the start and stop codons of the annotated gene model (pink model highlighted at bottom).
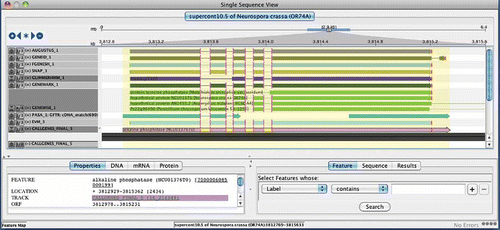
Figure 5. The Sybil Comparative Genomics Interface. A short region of synteny among orthologous genes of Aspergillus and related genomes is shown within the Sybil interface. Similarities and differences among the annotated gene structures become readily apparent, and many differences are found to represent artifacts rather than true evolutionary differences among related genes. Examples of the most striking discrepancies among annotated gene structures, involving different numbers of exons, or intron and exon lengths are highlighted by red rectangles.
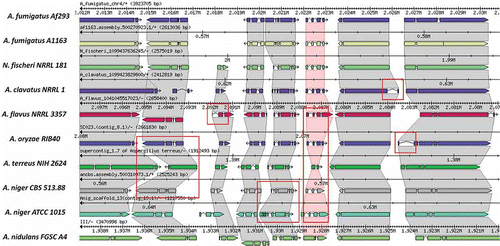
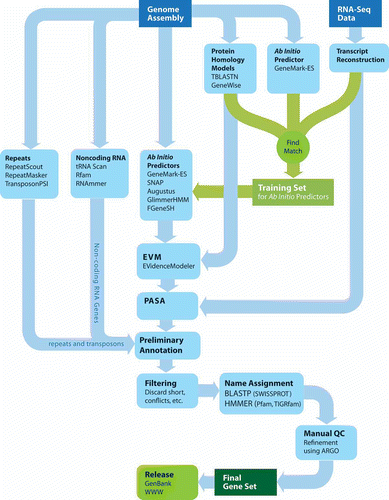
Figure 6. The Broad Institute Eukaryotic Genome Annotation Pipeline. Genome sequences are annotated by leveraging multiple sources of evidence for genes, including ab initio gene predictions, protein and transcript alignments, all of which are distilled into a consensus gene set. Gene products are named based on homology to proteins or domains of known function, manually refined, and ultimately released to public databases.