Figures & data
Figure 1. The developmental process of sequencing systems. First sequencing machine (370) was released by Applied Biosystems in 1986 and it led to the era of first generation sequencing. In 2005, Roche 454 entered market and it started the era of next generation sequencing. In 2006 Illumina's genome analyzer was born and SOLiD was released by Applied Biosystems in the following year.
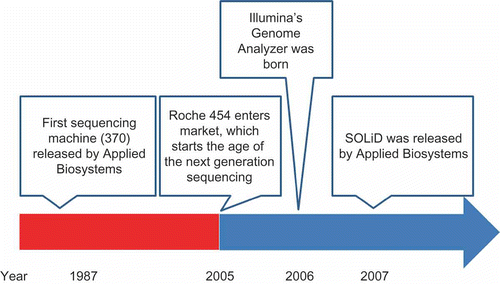
Table 1. Comparison of NGS platforms
Table 2. Comparison of the HiSeq and GA
Figure 2. The cost of human resequencing using GA and HiSeq. Price per 30× human genome is shown. In 2010, with HiSeq 2000, the cost per human genome dropped below $10K. It is expected that by the end of 2011, when HiSeq is upgraded, to yield 1T/run and the cost per human genome could reach $1000.
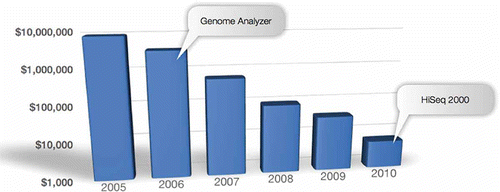
Table 3. Advantages of line imaging
Table 4. Error rate between GA and HiSeq
Figure 3. Amplication Plot in qPCR. Twenty different kinds of libraries with good sequencing data were chosen as standards, and the qPCR amplification curves of similar-type test libraries were compared to the standards for quantification. New standards were also compared to former ones, especially among dilutions when the 1-nM point was overlapped to guarantee overall stability of quantification QC.
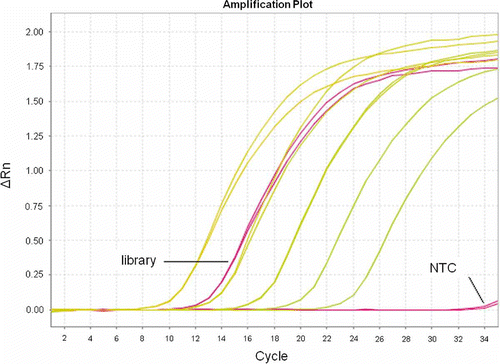
Figure 4. Standard curve in qPCR. Standard libraries were diluted to 1000, 100, 10, 1 and 0.1pM. It is required that at least four points are in the same line.
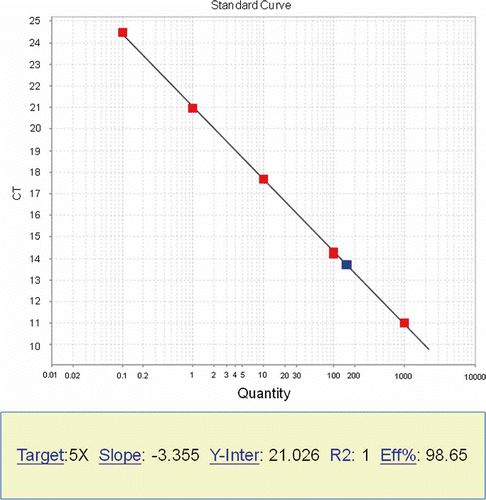
Table 5. Some problems detected in BGI and troubleshooting guide
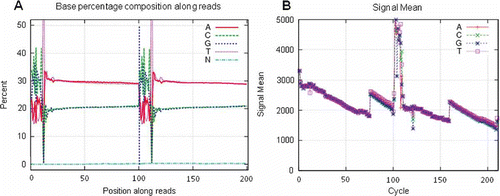
Figure 5. SQCS output. (A) Mate-pair library with abnormal small insert contamination may be due to incomplete digestion of linear DNA, insufficient biotinylated tag purification or nicked DNA genome. This could lead to assembly problems. (B) A 500-bp library with smear smaller contaminants. This may be caused by bad resolution of gel or gel contamination. (C) Abnormal base composition. Reasons may be complex genome or expression, such as Chinese shrimp, library preparation (inrandom fragmentation or ligation, adapter contamination, etc), or sequencing. (D) Abnormal base fluctuation due to adapter contamination.
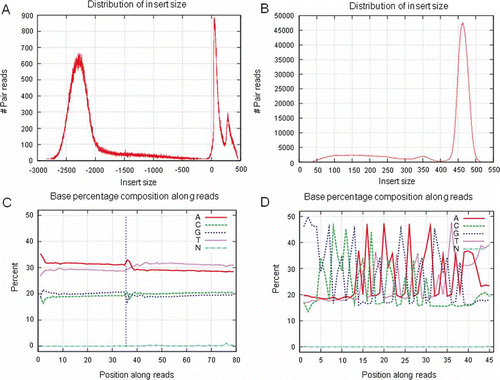
Figure 6. BGI's BMS system QC webpage.
Table 6. Fast QC check cluster and PF comparison between RTA and unmodified pipeline
Figure 7. Dark-area FC. (A) Pheonotype as wavy-edge dark area up to 50% of the lane. (B) Phenotype as straight-line and irregular dark area on one end of the lane. This could also lead to sequencing quality problems as low PF, bad focus, low Q20 and high error rate, etc.
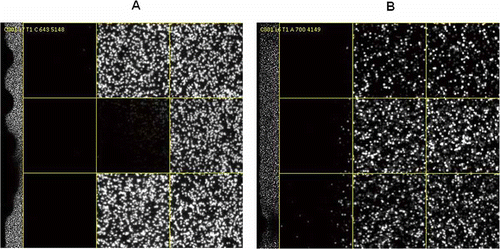
Table 7. A typical FC with gradient cluster densities
Table 8. Rate of base-calling duplication for different insert-sizes
Table 9. The duplicates in different HiSeq software versions
Figure 8. HiSeq quality data at different cluster densities. With increase in cluster density, the PF, Q20 and map rate decreased and mismatch rate increased. Quality has to be considered at the PF turnover point and before reaching the maximum yield and cluster density. In this figure, a density of around 700K/mm2 is critical for maximum yield.
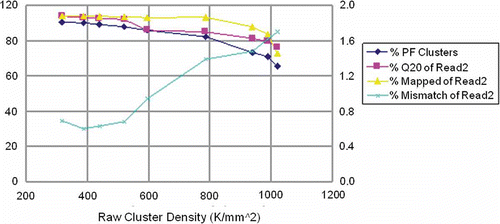
Table 10. HiSeq data with different cluster densities
Figure 9. Depth distribution versus GC content corresponding to . The genome was split into 1-kbp windows. Depth of sequences and GC was shown in each window. With an increase in cluster density, the GC content became lower and the GC bias becomes a problem. At high density in Lane F and I, GC distribution was abnormal.
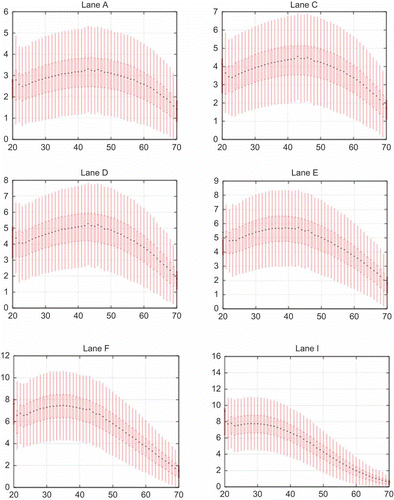
Figure 10. Correlation of GC content and genome base depth in Mycobacterium tuberculosis. The horizontal axis is GC content and the vertical axis is depth of gene base. (A) Truseq v3 reagent. (B) Truseq v2 reagent. Each data point corresponds to the number of reads recorded for a 500-bp window.
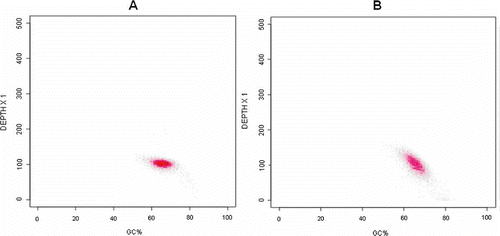
Figure 11. Improvement in sequencing quality by pooling more index libraries. Index library A shown above corresponds to