Figures & data
Figure 1. RNA processing in skeletal muscle. (a). Cross-section of a representative skeletal muscle. Individual multinucleated muscle fibers (blue dots) are bundled into fascicles. Each muscle fiber is made of myofibrils that contain the myofilaments, actin and myosin. T-tubules penetrate the muscle fiber and come into close proximity to the sarcoplasmic reticulum which surround the myofilaments. (b). Interactive view of RNA processing. After RNA polymerase-II (RNA Pol-II) transcribes DNA, newly synthesized mRNA is capped, cut and polyadenylated by the cleavage and polyadenylation factor, and then spliced. (c). Alternative splicing of a cassette exon that can either be included or skipped in the final mRNA transcript. (d). Alternative polyadenylation site selection. The 3ʹUTRs of some genes contain multiple polyadenylation signals that can be recognized by the polyadenylation and cleavage factor (CPSF). After the cleavage and polyadenylation factor binds to a polyadenylation signal (PAS), the polyA polymerase (PAP) adds the string of adenosines to the end of the mRNA transcript. This panel shows an example where three mRNA transcripts are generated by different polyadenylation site selection.
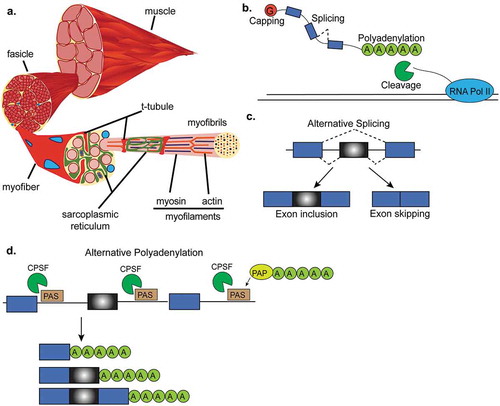
Figure 2. Regulation of mRNA capping and translation initiation by mTORC1. In response to anabolic stimuli, mTORC1 phosphorylates multiple downstream targets to govern the formation of the multi-protein eukaryotic initiation factor 4F complex (eIF4F) and subsequent translation initiation. The eIF4F complex is comprised of eIF4E (m7G mRNA cap binding protein), eIF4G (initiation factor scaffold protein), and eIF4A (RNA helicase) and its formation is the rate limiting step in translation initiation. Phosphorylation of eIF4E binding proteins 1 and 2 (4EBP1 and 4EBP2) frees eIF4E and allows it to bind to the m7G cap. The kinase activity of the 70 kDa ribosomal S6 protein kinase (S6K1) is increased in response to phosphorylation by mTORC1. S6K1 phosphorylates programmed cell death-4 (PDCD4) and relieves its inhibitory effect on eIF4A. S6K1 also phosphorylates eIF4B which further promotes helicase activity of eIF4A.
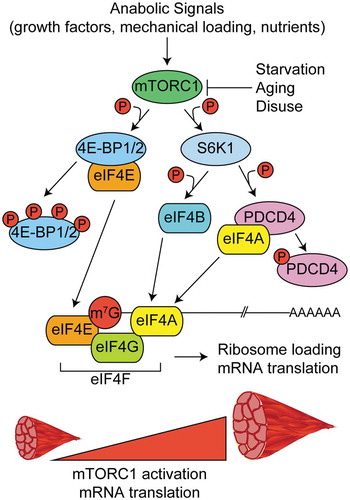
Figure 3. Regulation of alternative splicing in muscles. Several genes that are alternatively spliced produce different phenotypes in muscle depending on whether the alternative exon is included or skipped. (a). Alternative splicing of exon 11 of Bin1 pre-mRNA contributes to T-tubule formation and maintenance. (b). Skipping of exon 29 in Cav1.1 results in weak voltage sensitivity in adult skeletal muscle. (c). Inclusion of exon 11 of Insr pre-mRNA allows for stronger binding of insulin by the insulin receptor and thus insulin sensitivity. Skipping of exon 11 in adult muscles results in insulin resistance. (d). A switch from the adult to the fetal splicing isoforms of Ppp3ca, Ppp3cb and Ppp3cc leads to higher calcium phosphatase activity. (d). A switch in the splicing isoform of Cltc, Tmed2, Trip10 or Snap23 contributes to muscle homeostasis and T-tubule maintenance.
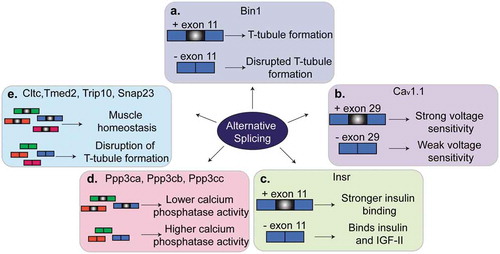
Figure 4. Regulation of polyadenylation in muscles. (a). The length of Pax3 3ʹUTR determines whether the transcript can be silenced by the microRNA miR-206. When a proximal polyadenylation site is used, the 3ʹUTR is shorter and lacks the miR-206 binding site and, in consequence, the transcript can escape the miR-206-induced silencing. In contrast, when a distal polyA site is used, the 3ʹUTR contains those binding sites leading to the silencing. (b). The polyA binding protein-1 (PABPN1) suppresses weak polyadenylation and cleavage sites by blocking the cleavage and polyadenylation factor (CPSF) from binding to these sites. Therefore, PABPN1 promotes the usage of a distal polyA site and thus longer transcripts by this mechanism. (c). Triple knockout of MBNL proteins in mice leads to the reactivation of fetal polyadenylation programs that preferentially use distal sites and thus generates short 3ʹUTRs. MBNL proteins bind similar sites as the CPSF does. In this way, binding of MBNL proteins close to the proximal polyA sites prevents CPSF from binding and promotes the selection of more distal polyA sites.
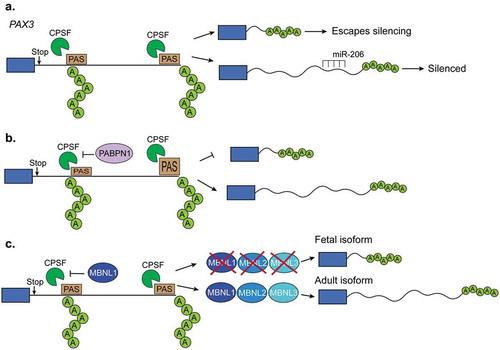
Figure 5. Expression of RNA-binding proteins during muscle development and myogenesis. The expression levels of several RNA-binding proteins (RBPs) change during muscle cell differentiation (myogenesis) (a) and skeletal muscle development (b). CELF: CUGBP Elav-like family member, MBNL: muscleblind like protein, QK: quaking, PTBP: polypyrimidine binding proteins, RBFOX: RNA binding fox-1 homolog.
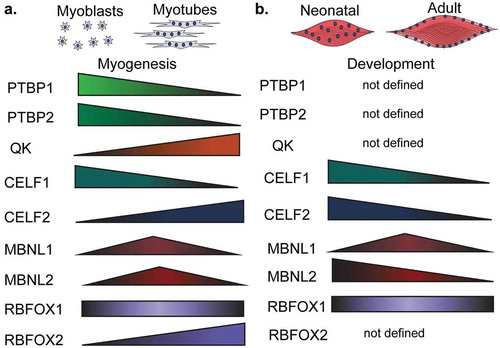
Figure 6. Regulation of myogenesis by MyoD1 activation of muscle genes. In myoblasts, MyoD1 associates with histone deacetylases (HDACs), leading to repression of muscle genes. During differentiation, MyoD1 interacts with histone acetyl transferases (HATs) to deposit acetylation (Ac) on histones of MyoD1 target genes. Acetylation of these muscle target genes promotes muscle fate determination.
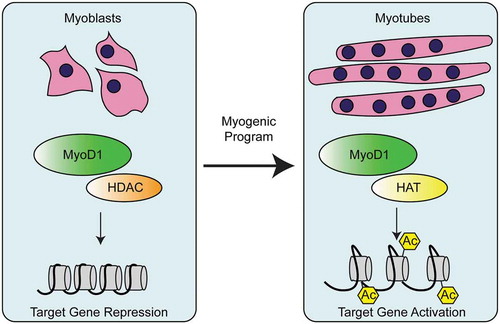
Figure 7. Myotonic dystrophy. (a). Myotonic dystrophy type-1 (DM1) is characterized by 50–3,000 CUG-repeats in the 3ʹUTR of DM1 protein kinase (DMPK) gene. The CUG-repeats cause RNA hairpins that sequester muscleblind like protein-1 (MBNL1) which leads to global changes in alternative splicing, polyadenylation, and mRNA stability. In DM1, the CUGBP Elav-like family member-1 (CELF1) is hyperphosphorylated and this leads to its gain-of-function. (b). Myotonic dystrophy type-2 (DM2) is characterized by 75–11,000 CCUG-repeats in the intron 1 of the CCHC-type zinc finger nucleic acid binding protein (CNBP, also known as ZNF9) gene. CCUG-hairpins sequester RNA binding fox-1 homolog proteins (RBFOX) which leads to extensive changes in alternative splicing and polyadenylation. CELF1 also becomes highly expressed and hyperphosphorylated in DM2. (c). Therapies for DM1. First, antisense oligonucleotides (ASOs) are used to block the transcription of portions CUG-repeats and in this manner MBNL sequestration is corrected. Second, gapmers that bind upstream of the CUG-repeats recruit RNAse-H which degrades the CUG-repeats and corrects the molecular defects of the disease. A third therapy is not RNA-based and instead uses various small molecules that block MBNL1 sequestration. This prevents the global misregulation of alternative splicing programs that occurs in DM1. (d). Congenital myotonic dystrophy (CDM) is characterized by 750–1,000 CUG-repeats at the 3ʹUTR of DMPK and hypermethylation of CpG islands upstream of the repeats. This results in sequestration of MBNL proteins as in DM1 and therefore, misregulation of alternative splicing, polyadenylation, and mRNA stability. Severe CDM results in muscle immaturity due to the improper activation of the cytokine, IL-6, an inhibitor of myoblast differentiation.
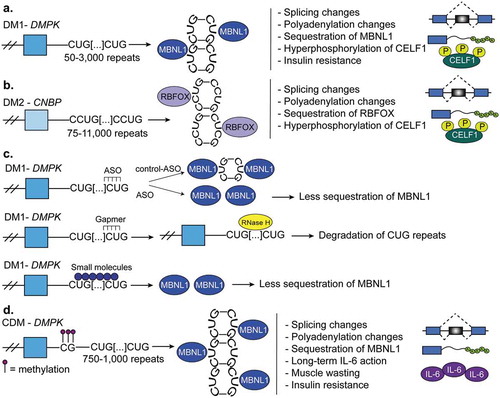
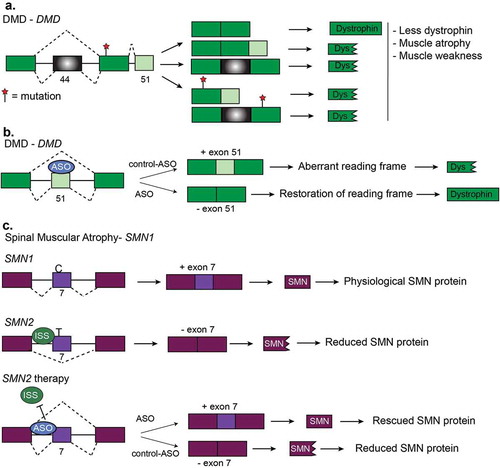
Figure 8. Other diseases and RNA-based therapies. (a). Duchenne muscular dystrophy (DMD) is characterized by mutations which disrupt the reading frame resulting in truncated dystrophin proteins. Truncated dystrophin leads to muscle atrophy and muscle weakness. Two frequent frameshift mutations are in exon 44 and exon 51. (b). Restoration of the DMD reading frame is possible using antisense oligonucleotides (ASOs). Here, ASOs are used to induce the skipping of exon 51 of DMD pre-mRNA. Skipping of this exon restores the reading frame and results the expression of functional dystrophin protein. (c). Generation of functional SMN protein in Spinal Muscular Atrophy (SMA). SMA is caused by a lack of survival motor neuron (SMN) protein. This happens when there is a deletion or mutation in the survival motor neuron-1 (SMN1) gene or increased copy number of SMN2. SMN2 produces much less functional SMN protein because of a C/T transition that prevents inclusion of exon 7. Multiple therapies have been devised for SMA that corrects for the loss of SMN1 by promoting exon inclusion in SMN2 via ASOs to increase SMN protein levels.