Figures & data
Figure 1. PIT is a superior proteomic method for detecting proteins derived from mobile genetic elements. (A) ‘Standard’ workflow for global proteomic analysis. Purified proteins are separated by gel electrophoresis, tryptically digested into <20-residue peptides, and analyzed by LC-MS/MS. Detected peptides are identified from their respective spectra using predicted proteins, peptides, and spectra ultimately derived from genome annotation and/or transposons reference databases. (B) PIT workflow. Experimental samples are split into protein (processed as described in A) and RNA; the latter is subjected to RNA sequencing. Short RNA-Seq reads are bioinformatically assembled into transcripts de novo (without referring to a reference genome),Citation49 and proteins, peptides, and spectra are predicted from these transcripts to produce a bespoke reference database for identifying peptides. In A & B, solid arrows indicate ‘wet’ experiments; dashed arrows indicate ‘dry’ (bioinformatic) analyses. (C) Bioinformatic identification of transposable elements expressing protein, in this case ‘transposon A’ (LTR retrotransposon used for illustration purposes). (i) Short peptides detected by LC-MS/MS often match multiple transposons sharing short stretches of perfect amino acid conservation, and individual elements deviate from the database consensus, complicating the assignment of peptides to their correct transposon. (ii) In PIT, detected peptides perfectly match their experimentally verified transcripts, allowing protein-producing mobile genetic elements to be accurately identified. When combined with a perfectly matched genome sequence, the precise genomic location of protein-producing transposons can also be determined (see ).
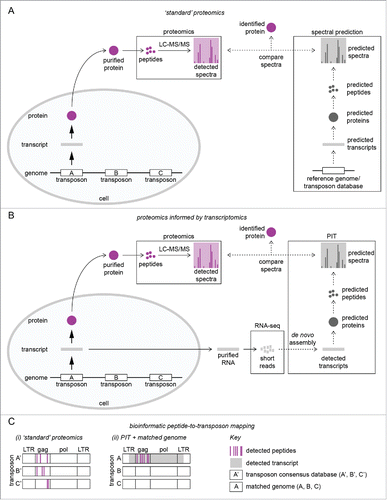
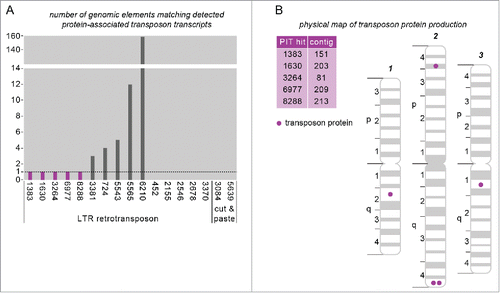
Figure 2. PIT can identify the specific unique genomic copy of an element that is expressing protein. (A) Number of genomic transposon sequences that precisely match PIT transcripts associated with 2 or more peptides. Transcripts with only one genomic match (purple bars) can be mapped to their exact source. Numbers (x axis) correspond to PIT hit IDs from our associated paper.Citation2 (B) Physical chromosome map for Ae. aegyptiCitation48; protein-expressing transposons for which the genomic source could be identified are annotated.