Figures & data
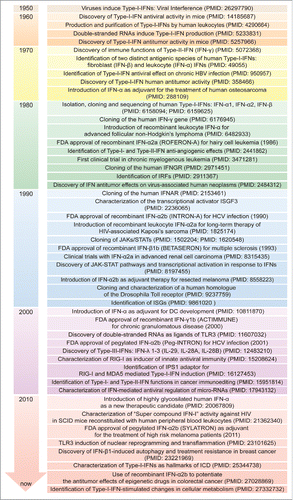
Figure 1. Major intracellular pathways leading to Type-I-IFN production. Families of sensors, known as PRRs, are available in the cells to detect viral and danger products, and induce the expression of Type-I-IFNs. One set of PRRs is localized in endosomal vesicles, while another set senses components in the cytoplasm. The endosome-associated TLR3 and the cytosolic MDA5, RIG-I and NOD2 sense double-stranded and single-stranded RNAs through the activation of adaptor molecules such as TRIF and MAVS, respectively. TRIF and MAVS in turn converge to activate the TBK1-IKKϵ kinase complex. This culminates in the activation of the transcription factors IRF3 and IRF7, which translocate to the nucleus and participate in the induction of a first wave of IFN-β production (1). IFN-β in turn acts in an autocrine/paracrine manner binding to the heterodimeric receptor IFNAR1-IFNAR2. This is followed by the activation of a JAK-STAT signaling pathway leading to a second wave of IFN-α production as well as to the transcription of other antiviral genes (2). Other PRRs sensing DNA are DAI and cGAS, with this last catalyzing the formation of ligands for STING upstream of the TBK1-IKKϵ complex, which finally drives the expression of IFNA and IFNB. cGAMP, cyclic guanosine monophosphate-adenosine monophosphate; cGAS: cyclic GMP-AMP synthase; DAI: DNA-dependent activator of IRFs; IFNs: interferons; IFNAR: IFN-α/β receptor; IKKϵ: IkB kinase ϵ; IRF: IFN regulatory factor; ISGs: IFN-stimulated genes; JAK: Janus kinase; MAVS: mitochondrial antiviral signaling adaptor; MDA5: melanoma differentiation-associated protein 5; NOD2: nucleotide oligomerization domain 2; PRRs: pathogen recognition receptors; RIG-I: retinoic acid-inducible gene-I; STAT: signal transducer and activator of transcription; STING: stimulator of IFN genes; TBK1: TANK-binding kinase 1; TLR3: Toll-like receptor 3; TRIF: TIR-domain containing adaptor protein-inducing IFN-β; TYK2, tyrosine kinase-2.
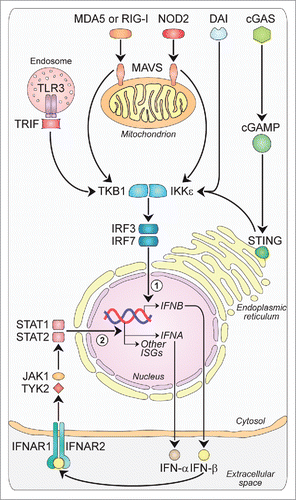
Figure 2. Type-I-IFN-triggered signals. Type-I-IFNs may favor tumor regression and/or tumor progression by acting on tumor cells, immune cells and endothelial cells via various mechanisms. First, acting on tumor cells Type-I-IFNs may promote either tumor regression, by inducing cell-cycle arrest (1), apoptosis (2) and enhanced immunogenicity through cell surface expression of MHC-I (3) and TAAs (4), or tumor progression by inducing resistance to apoptosis (5), EMT (6), tumor-cell stemness (7), and the upregulation of immune-inhibitory signals such as PD-L1 (8). Second, acting on the vascular and lymphatic system Type-I-IFNs inhibit angiogenesis through VEGF downregulation (9). Finally, acting on the immune system Type-I-IFNs may favor tumor regression by stimulating the maturation of DCs (10), promoting the release of pro-inflammatory cytokines (11), favoring CTL cross-priming (12), fostering the activation and survival of CD8+ and CD4+ T cells (13) and of NK cells (14), having a crucial role on core energetic metabolism regulation (15), and negatively regulating immune suppressive Treg cells (16) and MDSCs (17). CSC: cancer stem cell; DC: dendritic cell; EMT: epithelial-to-mesenchymal transition; IFNs: interferons; MDSC: myeloid-derived suppressor cell; MHC-I: major histocompatibility complex-I; NK: natural killer; PD1: programmed death 1; PD-L1: programmed death-ligand 1; TAAs: tumor-associated antigens; TCA: tricarboxylic acid; Treg: regulatory T cell; VEGF: vascular endothelial growth factor; VLS: vascular and lymphatic system.
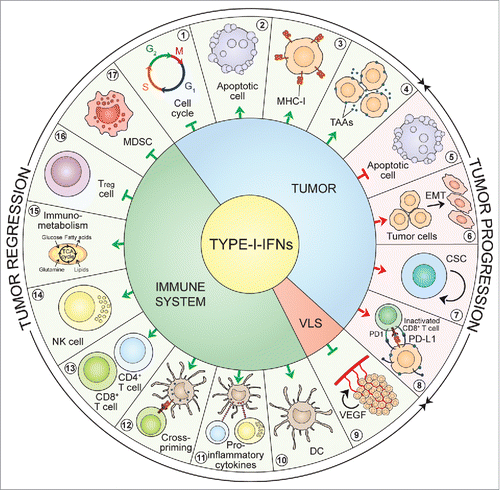
Figure 3. Timeline of IFN discovery and clinical use. The discovery of IFNs evolved from studies of viral interference beginning in 1950. Since then, great attention has been devoted to the molecular understanding and clinical use of IFNs for virus-related and -unrelated malignancies. DC: dendritic cell; FDA: Food and Drug Administration; HBV: hepatitis B virus; HCV: hepatitis C virus; HIV: human immunodeficiency virus; ICD: immunogenic cell death; IFNs: interferons; IFNAR: IFN-α/β receptor; IFNGR: IFN-γ receptor; IPS-1, IFN-beta promoter stimulator-1; IRF: IFN regulatory factor; ISG: IFN-stimulated gene; ISGF3: IFN-stimulated gene factor 3; JAK: Janus kinase; MDA5: melanoma differentiation-associated protein 5; RIG-I: retinoic acid-inducible gene-I; SCID: severe combined immunodeficiency; STAT: signal transducer and activator of transcription; TLR3: Toll-like receptor 3.