Figures & data
Figure 1. Strategy to identify MC-38 colorectal tumor specific neo-peptides. MC-38 DNA was isolated and its sequenced exome was compared to DNA from C57BL/6 wild-type mice for genetic variants. Corresponding amino acid sequences were used to predict potential binders to MHC class I Kb and Db by NetMHC4.0 prediction algorithm. A combination of immunoprecipitation of MHC class I from MC-38 lysate and subsequent mass-spectrometry of eluted peptides was performed to establish MHC class I presentation.
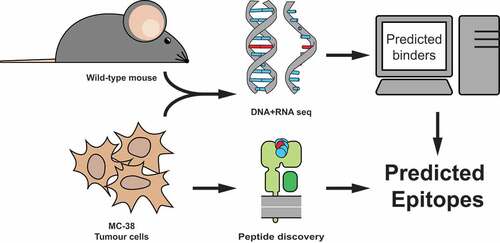
Table 1. Selected candidate neo-epitopes. All eight neo-peptides shown were selected by exome and RNA sequencing and confirmed with synthetic peptide mass-spectrometry for MHC class I presentation. IC50 of presented peptide affinity was predicted with NetMHC (ANN) for respective MHC alleles. Candidate neo-epitopes indicated with (*) were previously described in Yadav M. et al. (2014). Context of these candidate neo-epitopes was included in the form of variant frequencies in DNA and RNA, whole gene expression levels in MC-38 (in transcripts per million), and the observed presence of wild-type (WT) peptides in the elution.
Figure 2. Irradiated MC-38 immunization induces CD8+ T cells specific for candidate neo-epitopes. (a) Mice were prime-boost vaccinated with subcutaneous injection of 5 × 106 irradiated MC-38 tumor cells. (b) Cytokine production of CD8α+ T lymphocytes through stimulation with synthetic “long” peptide-loaded dendritic cells ex vivo and (c) after restimulation with irradiated MC-38 cells. (b) and (c) are from two independent experiments, n = 3 for each. Shown are representative cytokine (IFNγ and TNFα) staining plots from CD8α+ populations (left) and plotted percentages of double-positive IFNγ and TNFα cytokine producing populations of individual mice (right).
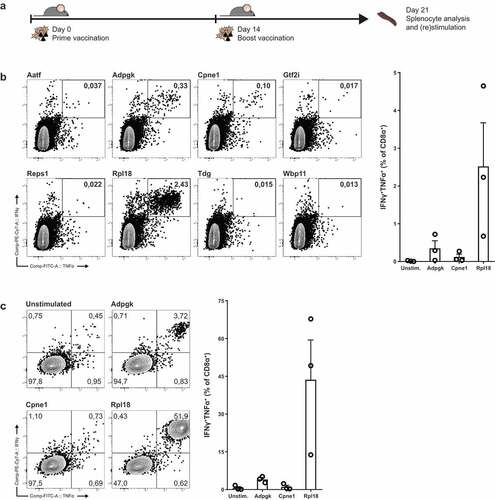
Figure 3. Tumor-bearing mice have circulating and tumor-infiltrating CD8+ lymphocytes specific for Rpl18 neo-peptide. (a) Mice were inoculated with 3 × 105 live MC-38 tumor cells. Lymphocytes isolated from spleen and tumor-infiltrating lymphocytes from the established tumors were stimulated with neo-peptides eight days post inoculation (n = 4). (b) IFNγ and TNFα cytokine production of CD8α+ T lymphocytes through stimulation with synthetic “long” peptides-loaded dendritic cells; from a single mouse (left) and all double-cytokine producing summarized (right). Where indicated, statistical significance was determined with student’s t test.
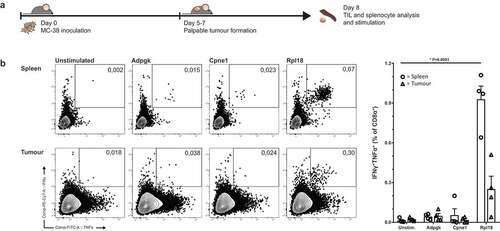
Figure 4. Dominant Rpl18-specific CD8+ populations in immunologically protected mice through PD-L1 checkpoint blockade therapy. (a) Mice bearing palpable tumors 6 days after inoculation with MC-38 tumor cells were injected thrice with anti-PD-L1 in a three-day interval to induce complete tumor control. At eighty days post inoculation, mice were re-challenged with MC-38 to confirm immunological protection. Tumor-specific T cells were expanded through co-culture with irradiated MC-38 tumor cells for one week and analyzed by peptide-loaded dendritic cell stimulation o/n (n = 6). (b) A shortened treatment schedule was used to analyze Rpl18-specific T cells in mice during tumor regression (left panel). At twelve days post challenge, lymphocytes specific for Rpl18 neo-epitope were observed in spleens and LN’s through tetramer staining (middle panel) and ICS (right panel). Anti-PD-L1 treatment increased Rpl18-specific T cell frequencies in both lymphoid organs (n = 7, statistical significance was determined with student’s t test, representative results of two independent experiments).
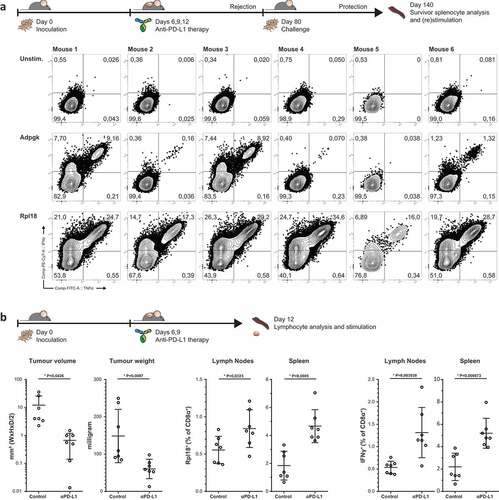
Figure 5. Specificity to mutated peptide and direct recognition of tumor cells. (a) Minimal synthetic neo-peptide was compared to N- and C-terminally extended “long” peptides in intracellular cytokine staining of activated T cells upon coculture with peptide-loaded dendritic cells. (n = 6, error bars depict SEM) (b) Rpl18-specific T cell bulks were stimulated with either mutated or wild-type extended peptides to determine specificity to the single amino-acid mutation. (c) In vitro recognition of live MC-38 vs B16F10 melanoma control tumor cells by tetramer-stained T cells in Rpl18+ (5 out of 6 original bulks) and Adpgk+ (3 out of 6) cultures. A representative gating-plot of a single culture is shown (top). Tumor-specific single and double cytokine production for both Adpgk and Rpl18-specific T cells is shown below.
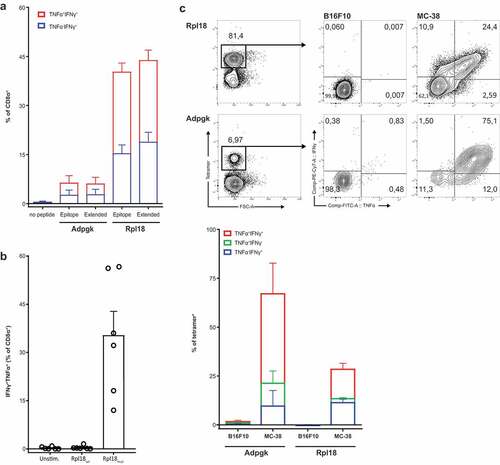
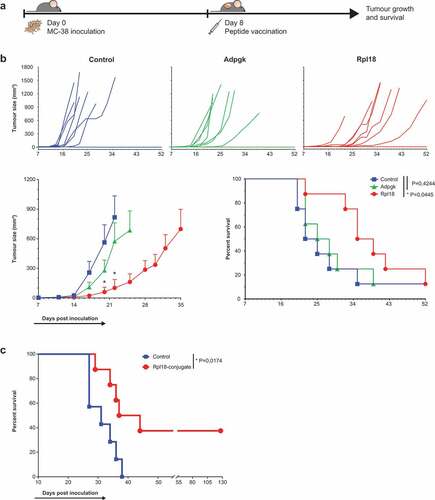
Figure 6. Therapeutic vaccination with Rpl18 neo-peptide-based vaccinations improves tumor control. (a) Schematic representation of therapeutic vaccination protocol. 3 × 105 tumor cells were subcutaneously injected in the right flank. Eight days post inoculation, tumors established and a single synthetic neo-peptide vaccination was given intradermally at the tail-base. (b) Therapeutic vaccination with 10 nmol of extended synthetic peptide harboring neo-epitopes of Adpgk or Rpl18 delayed outgrowth of MC-38 tumors. At day 20, average tumor size in Adpgk-vaccinated mice was two-fold reduced (279 vs. 563 mm3, P = 0,1894), while Rpl18 vaccination reduced tumor size nearly 10-fold (60 mm3, P = 0,0163). This trend was maintained 22 days post inoculation (Adpgk: 574 vs. 817 mm3, P = 0,4098; Rpl18: 102 vs. 817 mm3, P = 0,0083). Subsequent median survival was extended in Rpl18 vaccinated mice (37 vs 23,5 days, P = 0,0445), while no significant extension was observed for Adpgk-vaccinated mice (26,5 days, P = 0,4244). (Eight mice per group; error bars indicate SEM; multiple t tests and Gehan-Breslow-Wilcoxon test were used for statistical analysis of tumor size and survival, respectively) (representative results of two independent experiments). (c) To improve vaccination efficacy we conjugated the Rpl18 neo-peptide to Upam TLR2-ligand (also known as Amplivant®). Therapeutic vaccination with 10 nmol of Rpl18-conjugate against established tumor significantly improved tumor control and survival compared to Upam-control (P = 0,0174; n = 8 vs n = 7 in control group; log rank tested) (depicted results are from a single experiment).