
Figures & data
Figure 1 Schematic representation of SCN4A mutations identified in ALS patients. Clinically reported neuromuscular phenotype highlighted in boxes. Identified genetic changes within SCN4A are proposed to lead to motor neuron toxicity by one of two mechanisms: either directly via excessive membrane sodium permeability leading to hyperexcitability and ultimately excitotoxicity; or indirectly via muscle hyperexcitability leading to retrograde motor neuron toxicity.
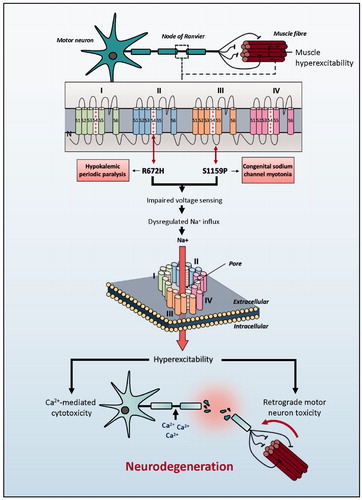
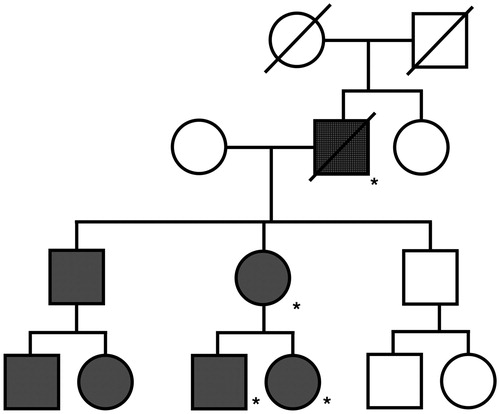
Figure 2 Pedigree from index case. Pedigree showing co-segregation of SCN4A p.Ser1159Pro with congenital myotonia. (shaded: congenital myotonia, crosshatch: ALS and congenital myotonia, * = SCN4A p.Ser1159Pro mutation confirmed).
Supplemental material