Figures & data
Figure 1. The dystrophin and utrophin-associated protein complexes and utrophin based therapies. (a) In healthy muscle, the dystrophin and its associated proteins (DAPC) protect the sarcolemma from contraction-induced injury, maintaining the biomechanical properties of fibre strength, flexibility and stability in skeletal muscle. Dystrophin contains N-terminal (NTD), central rod, cysteine-rich (CRD), and C-terminal (CTD) domains. The NTD and spectrin-like repeat 11–15 bind the costameric actin filament to aid in shock absorbance during muscle contraction. The central rod domain contains 24 spectrin-like repeats and 4 putative hinge modules. Spectrin repeat 11–17 interact with a second independent actin binding domain and spectrin repeats 16/17 in dystrophin are able to recruit nNOS. Dystrophin interacts indirectly with microtubules through ankyrin-B and directly via spectrin repeats 20–23. The CRD links dystophin to the sarcolemmal-bound β-dystroglycan which binds to α-dystroglycan to form the dystroglycan complex. β-dystroglycan in association with δ-sarcoglycan participates in the stabilization of the (α, β, δ and γ) sarcoglycan-sarcospan complex at the sarcolemma and the α-dystroglycan links laminin-α2 at the extracellular matrix which both strengthen the DPAC. The CTD binds several cytosolic proteins such as α-dystrobrevin 2, α- and β-syntrophins and recruits signalling molecules such as nNOS via PDZ domains. (b) At the neuromuscular junction, utrophin forms the UAPC complex with similar protective functions as the DAPC. Acting as a receptor for agrin and laminin, the UAPC links the actin cytoskeleton through the NTD to the extracellular matrix via laminin-α4, -α5, and -β2. Utrophin binds to MAST and is associated with microtubules by the CTD and nNOS can only be recruited indirectly through the syntrophins. In addition, the UAPC binds to rapsyn and is involved in the clustering of acetylcholine receptors. (c) In DMD, the absence of dystrophin results in loss of the DAPC w hich lead to costamere perturbation and sarcolemma fragility. Subsequent sarcolemmal lesions trigger muscle degeneration and chronic inflammation associated with failed regeneration leading to necrotic and fibrotic events. (d) As a structural and functional surrogate to dystrophin, utrophin can replace and compensate for the lack of dystrophin in dystrophic muscles. A large majority of the elements of the DAPC and in consequence sarcolemma stability is rescued leading to prevention of the dystrophic phenotype despite the lack of restoration of the microtubules and nNOS.
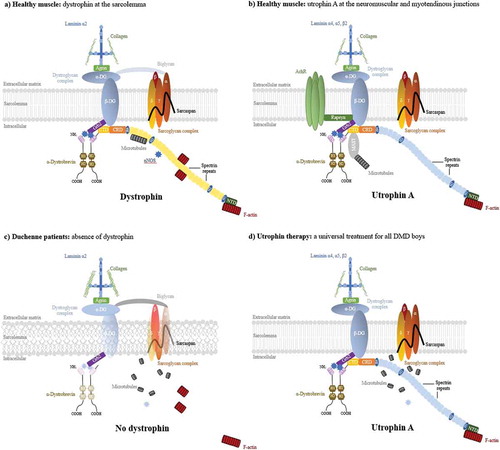
Figure 2. Schematic organisation of the dystrophin (A), utrophin (B) genes with associated promoters and utrophin A and B transcripts (c). (a). Dystrophin expression is driven by at least 7 tissue-specific promoters. The full-length dystrophin transcription is controlled by 3 promoters (B, brain; M, muscle; P, Purkinje) localised upstream to the first exon and which only differs in its NH2-terminal sequences. 4 internal promoters, dp260, dp140, dp116 and dp71 were reported. Dp427m is the main variant produced in muscle and is involved in DMD and BMD. (b). In contrast to dystrophin, the utrophin gene has a long 5ʹprime untranslated region composed of 2 exons and a cluster of unmethylated, rare-cutting restriction enzyme sites. Expression of utrophin transcripts is controlled by two independently regulated promoters, A and B. Whereas each promoter results in a transcript with unique 5ʹ exons that splice into a common mRNA at exon 3 (black box), utrophin A promoter is CpG rich at the 5ʹ-end and contains an N-box motif controlling the synaptic expression. Different elements of the utrophin A promoter are specified. Promoter B, devoid of the N-box motif lies within the second exon of utrophin. 3 shorter isoforms Up140, G-Utrophin and Up71 were reported similar to the smaller transcripts of the dystrophin gene. Exons (grey boxes with exon numbers) and intronic regions (black line), intronic enhancer (DME-1 for dystrophin, DUE for utrophin, green boxes), untranslated first exon 1A of utrophin (yellow) are specified. Arrows indicate transcription start sites. (c) Schematic representation of the utrophin-A and B transcripts. The unique translated exon of the utrophin A transcript is 2A. Utrophin B transcript starts at exon 1B. Promoters A and B produce proteins that differ at their N-termini with, respectively, unique sections of 31 and 26 amino acids. Adapted from [Citation11]. Full color available online.
![Figure 2. Schematic organisation of the dystrophin (A), utrophin (B) genes with associated promoters and utrophin A and B transcripts (c). (a). Dystrophin expression is driven by at least 7 tissue-specific promoters. The full-length dystrophin transcription is controlled by 3 promoters (B, brain; M, muscle; P, Purkinje) localised upstream to the first exon and which only differs in its NH2-terminal sequences. 4 internal promoters, dp260, dp140, dp116 and dp71 were reported. Dp427m is the main variant produced in muscle and is involved in DMD and BMD. (b). In contrast to dystrophin, the utrophin gene has a long 5ʹprime untranslated region composed of 2 exons and a cluster of unmethylated, rare-cutting restriction enzyme sites. Expression of utrophin transcripts is controlled by two independently regulated promoters, A and B. Whereas each promoter results in a transcript with unique 5ʹ exons that splice into a common mRNA at exon 3 (black box), utrophin A promoter is CpG rich at the 5ʹ-end and contains an N-box motif controlling the synaptic expression. Different elements of the utrophin A promoter are specified. Promoter B, devoid of the N-box motif lies within the second exon of utrophin. 3 shorter isoforms Up140, G-Utrophin and Up71 were reported similar to the smaller transcripts of the dystrophin gene. Exons (grey boxes with exon numbers) and intronic regions (black line), intronic enhancer (DME-1 for dystrophin, DUE for utrophin, green boxes), untranslated first exon 1A of utrophin (yellow) are specified. Arrows indicate transcription start sites. (c) Schematic representation of the utrophin-A and B transcripts. The unique translated exon of the utrophin A transcript is 2A. Utrophin B transcript starts at exon 1B. Promoters A and B produce proteins that differ at their N-termini with, respectively, unique sections of 31 and 26 amino acids. Adapted from [Citation11]. Full color available online.](/cms/asset/5987e9c3-fa2e-40bb-abf3-71b2f0278f75/ieod_a_1438261_f0002_oc.jpg)
Figure 3. The domain composition of the full-length dystrophin protein (A), full-length utrophin A and B and various shorter isoforms (B). (a) Dystrophin contains an amino-terminal domain (NTD) followed by 24 spectrin like central rod domain, the cysteine rich (CRD) and the carboxy-terminal (CTD) domain. The full-length dystrophin proteins Dp427m, Dp427b and Dp427p are respectively expressed in skeletal, smooth and cardiac muscle, brain and Purkinje neuron. Dystrophin interacts with actin-F via two low affinity sites via the amino-terminal CH domain and spectrin repeats 11–17. Other binding sites are indicated. (b) The full-length utrophin A and B proteins present the same overall structure as the dystrophin full-length protein but have a shorter central rod domain and a 22 spectrin like domain. Utrophin is interacting with actin filaments through a unique continuous binding domain via the N-terminal domain and the spectrin 1–10. Utrophin A and B differ at their N-termini with unique sections of 31 and 26 amino acids, respectively. Various shorter utrophin isoform as Up140 and Up71 ubiquitously expressed were reporter as G-utrophin expressed in brain and sensory ganglia. Identity and similarity score per domain and expression patterns are specified. Data taken from [Citation6,Citation27].
![Figure 3. The domain composition of the full-length dystrophin protein (A), full-length utrophin A and B and various shorter isoforms (B). (a) Dystrophin contains an amino-terminal domain (NTD) followed by 24 spectrin like central rod domain, the cysteine rich (CRD) and the carboxy-terminal (CTD) domain. The full-length dystrophin proteins Dp427m, Dp427b and Dp427p are respectively expressed in skeletal, smooth and cardiac muscle, brain and Purkinje neuron. Dystrophin interacts with actin-F via two low affinity sites via the amino-terminal CH domain and spectrin repeats 11–17. Other binding sites are indicated. (b) The full-length utrophin A and B proteins present the same overall structure as the dystrophin full-length protein but have a shorter central rod domain and a 22 spectrin like domain. Utrophin is interacting with actin filaments through a unique continuous binding domain via the N-terminal domain and the spectrin 1–10. Utrophin A and B differ at their N-termini with unique sections of 31 and 26 amino acids, respectively. Various shorter utrophin isoform as Up140 and Up71 ubiquitously expressed were reporter as G-utrophin expressed in brain and sensory ganglia. Identity and similarity score per domain and expression patterns are specified. Data taken from [Citation6,Citation27].](/cms/asset/2e46230e-7810-4470-9e09-18013512d644/ieod_a_1438261_f0003_oc.jpg)
Figure 4. Dynamic utrophin protein levels in mdx dystrophic muscles. Between the age of 3 and 10 weeks, dystrophin deficient muscles of the mdx mouse undergo many degeneration and regeneration cycles which contribute to the progressive muscle wasting. The dynamic utrophin level is variable and dependent on the muscle, the age and the animal and associated with high regeneration and repair processes emphasizing the complexity of quantifying utrophin in dynamic and dystrophic muscles.
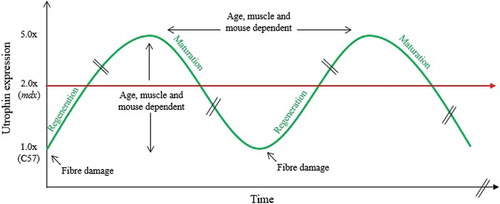
Table 1. Status of the main and most promising utrophin therapeutic strategies for DMD.
Table 2. Inclusion and exclusion criteria in PhaseOut DMD trial with ezutromid.
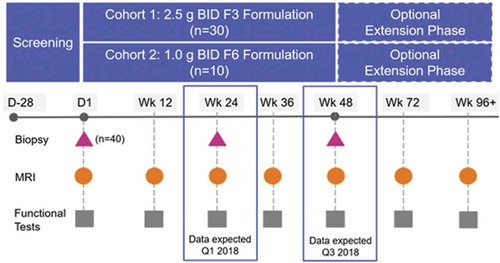
Figure 5. Overview of the PhaseOut DMD, a Phase 2 Clinical Trial of Ezutromid in Patients with DMD.