
Figures & data
Figure 1. Crystal structure of human c-Src kinase (PDB: 2SRC). c-Src is a monomeric protein consisting of three domains (a catalytic domain CD and two peptide-recognition domains SH3 and SH2) and two SBPs (polyproline-II helical peptide PPII and C-terminal phosphorylatable tail CTPT). Phosphorylation of Tyr527 residue (pTyr527) results in CTPT binding to SH2 domain, which further induces SH3–PPII interaction. Consequently, the CD domain is locked in an autoinhibitory form.
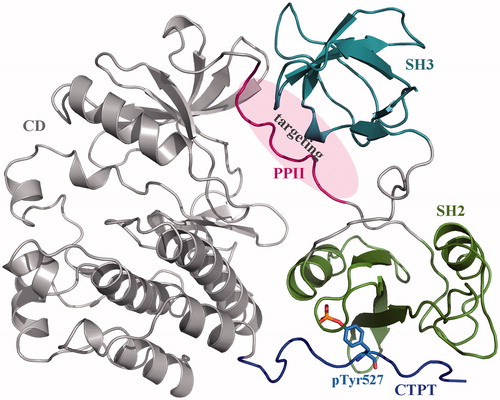
Figure 2. Superposition of MD-derived PPII structural snapshots. (a) The crystal structure of full-length c-Src protein was subjected to 100-ns MD simulations, and the structural snapshots of PPII segment in the full-length protein were saved at 0, 25, 50, 75 and 100 ns and superposed onto each other. (b) The PPII peptide was stripped from the c-Src crystal structure, (c) which was then subjected to 100-ns MD simulations, and its structural snapshots were saved at 0, 25, 50, 75 and 100 ns and superposed onto each other.
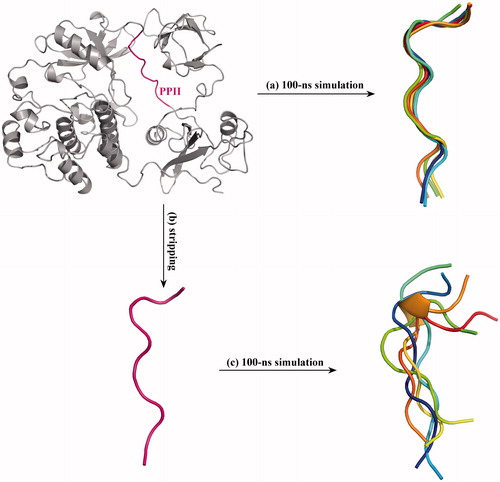
Table 1. The binding energetics and affinity of peptide ligands to c-Src SH3 domain.
Figure 3. Superposition of class I c-Src SH3–PLR2 complex crystal structure (PDB: 1PRL) (a) and class II c-Src SH3–PLR1 complex crystal structure (PDB: 1RLP) (b) onto the SH3–PPII adduct of c-Src kinase crystal structure (PDB: 2SRC). From the superposition the binding mode of PPII to SH3 domain is revealed as class II, where the three key residues Pro0, Pro3 and Arg5 of class II motif P0xxP3x+5 in PLR1 peptide can well match the residues Pro250, Gln253 and Leu255 of PPII, respectively.
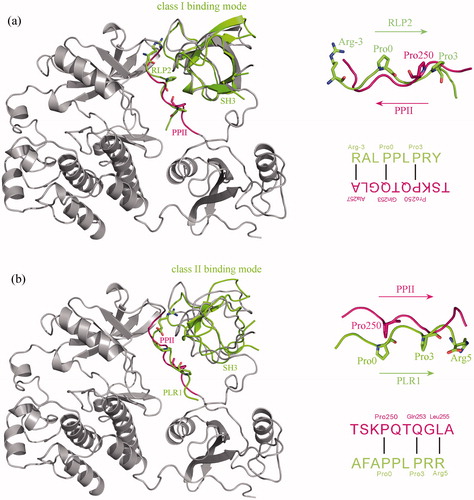
Figure 4. The intermolecular interaction between c-Src SH3 domain and PPII peptide. (a) The equilibrium conformation of c-Src SH3 domain in complex with PPII peptide. The residue positions that play different roles in the domain–peptide binding are highlighted in different colours. Magenta (Pro250, Gln253 and Leu255): key residues in the PxxPx + motif. Indigo (Lys249 and Thr252): the residues pointing to the domain pocket. Yellow (Gln251 and Gly254): the residues pointing out of the domain pocket. Green (Thr247 and Ser248): the two N-terminal residues that are far away from the domain. Hoar (Ala256): the C-terminal residue that interacts slightly with the domain. (b) Hydrogen bonds and hydrophobic interactions across the complex interface of c-Src SH3 domain with PPII peptide (produced using LigPlot program, Citation23]. (c) Computational alanine scanning of PPII peptide binding to c-Src SH3 domain. Positive and negative ΔΔGAla values indicate favourable and unfavourable residues in the domain–peptide binding, respectively.
![Figure 4. The intermolecular interaction between c-Src SH3 domain and PPII peptide. (a) The equilibrium conformation of c-Src SH3 domain in complex with PPII peptide. The residue positions that play different roles in the domain–peptide binding are highlighted in different colours. Magenta (Pro250, Gln253 and Leu255): key residues in the PxxPx + motif. Indigo (Lys249 and Thr252): the residues pointing to the domain pocket. Yellow (Gln251 and Gly254): the residues pointing out of the domain pocket. Green (Thr247 and Ser248): the two N-terminal residues that are far away from the domain. Hoar (Ala256): the C-terminal residue that interacts slightly with the domain. (b) Hydrogen bonds and hydrophobic interactions across the complex interface of c-Src SH3 domain with PPII peptide (produced using LigPlot program, Citation23]. (c) Computational alanine scanning of PPII peptide binding to c-Src SH3 domain. Positive and negative ΔΔGAla values indicate favourable and unfavourable residues in the domain–peptide binding, respectively.](/cms/asset/941e1ea4-2936-449a-8115-380a6403bce8/ianb_a_1360327_f0004_c.jpg)
Table 2. The binding energetics and affinity of PPII-derived peptides to c-Src SH3 domain.
Figure 5. The mutation energy profile of c-Src SH3–peptide interaction. The mutation energies (ΔΔG) are coloured by binding energy change upon PPIIm5 peptide residue mutation. (a) Systematic single-point mutation profile of the anchor residues Lys249 and Thr252 of PPIIm5 peptide, where each residue was mutated to all other 19 amino acid types. (b) Favourable double-point mutation profile of the anchor residues Lys249 and Thr252 of PPIIm5 peptide, where the mutation only consider the combination of four favourable amino acids Y, F, I and L at residue 249 and three favourable amino acids P, I and L at residue 252.
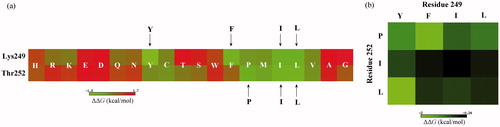
Figure 6. Comparison between the nonbonded interaction patterns across the complex interfaces of c-Src SH3 domain with PPII and PPIIm7 peptide ligands. The nonbonded interactions were identified using PLIP server [Citation27].
![Figure 6. Comparison between the nonbonded interaction patterns across the complex interfaces of c-Src SH3 domain with PPII and PPIIm7 peptide ligands. The nonbonded interactions were identified using PLIP server [Citation27].](/cms/asset/31ab0508-aab1-449e-a768-91263c273481/ianb_a_1360327_f0006_c.jpg)