Figures & data
Figure 1. The optimized structure and numbering of the most stable and unstable of TT conformers.
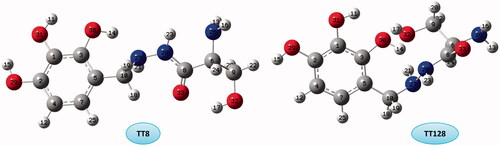
Figure 2. The optimized structure and numbering of the most stable and unstable of CC conformers.
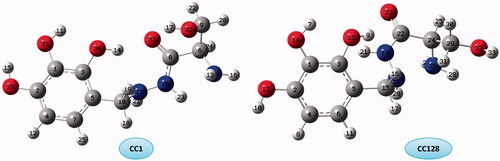
Figure 3. The optimized structure and numbering of the most stable and unstable of TC conformers.
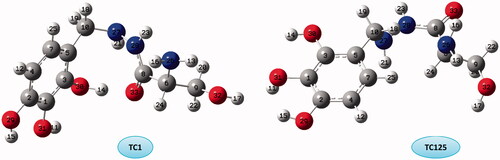
Figure 4. The optimized structure and numbering of the most stable and unstable of CT conformers.
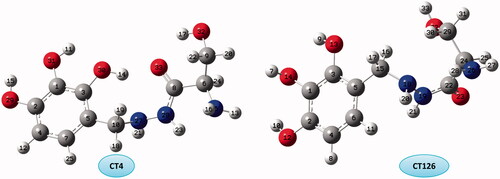
Figure 5. The molecular graphs of CT4 obtained from the B3LYP/6–31 G(d,p) wave function.
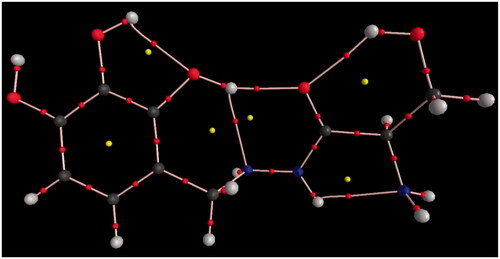
Table 1. The geometrical parameters (bond distance in Å) and topological parameters (in a.u.) of in more stable conformers.
Table 2. Topological parameters (in a.u.) and intramolecular hydrogen bond energies (in kJ/mol) of stable and unstable conformers of benserazide.
Table 3. The NBO analysis of stable and unstable conformers of benserazide (includes the second order perturbation energy of some important orbital interactions).
Table 4. Total energy, dipole moment, and MO analyses of stable and unstable conformers of benserazide.
Table 5. The harmonic stretching vibrational frequencies of O…H and N…H for the most stable conformations of benserazide.
Figure 6. Optimized geometry of carboxylated carbon nanotube (a) from front and (b) from side view.
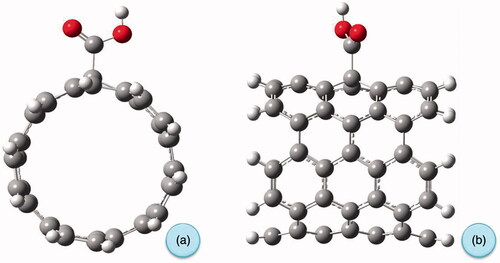
Figure 7. Non-covalent adsorption features of benserazide onto carboxylated carbon nanotube.
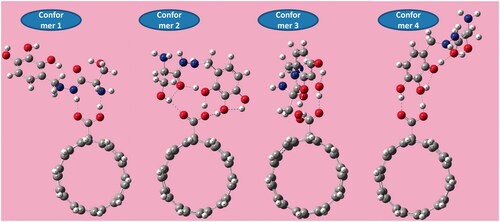
Figure 8. Molecular graphs of the conformers calculated at B3LYP/6–311 G(d) level using AIM approach.
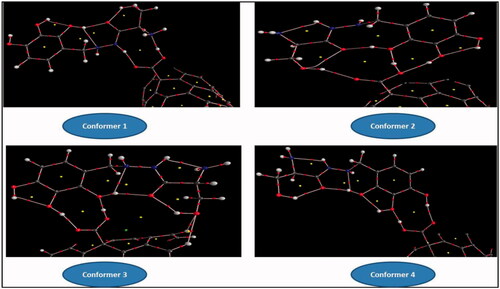
Table 6. Adsorption energy, Eads in kJ/mol, optimum distance of interaction, in Ǻ, electron density (ρBCP), Laplacian (∇2ρBCP), electron kinetic energy density (GBCP), electron potential energy density (VBCP), total electron energy density (HBCP) and hydrogen bond energy (EHB) at the bond critical point (BCP) of all configurations calculated at B3LYP/6–311 G(d) level.
Figure 9. Frontier molecular orbitals (HOMO-LUMO) plots with corresponding energy gap of (a) carboxylated carbon nanotube and (b) benserazide molecule.
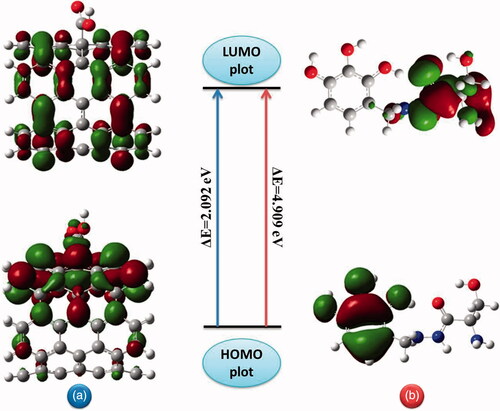
Figure 10. The pictorial illustration of HOMO and LUMO frontier molecular orbitals with corresponding energy gap for different possible complexes calculated by B3LYP method with 6–311 G(d) basis set; (a) conformer 1, (b) conformer 2, (c) conformer 3 and (d) conformer 4.
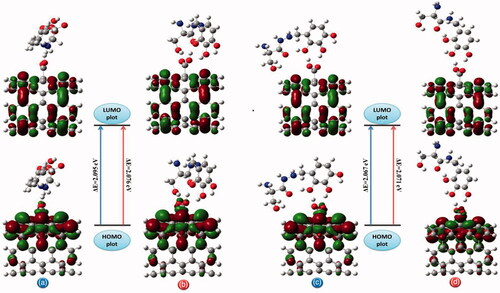
Table 7. Total energy (Hartree) and global reactivity descriptors (all units in eV) for BZ/c-CNT drug carrier conformers and its constituents.
Figure 11. MPE plots of (a) Benserazide, (b) c-CNT, (c) conformer 1, (d) conformer 2, (e) conformer 3 and (f) conformer 4 calculated at B3LYP/6–311 G(d).
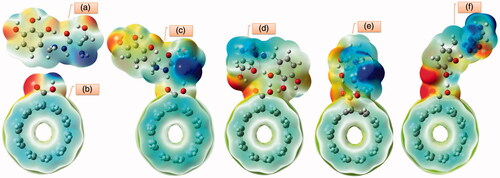
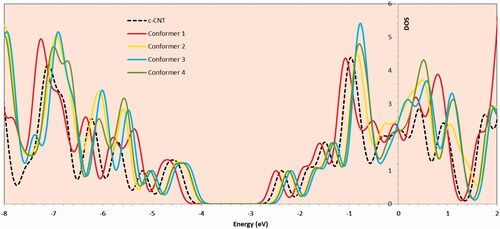
Figure 12. Total DOS of the c-CNT and Bz/c-CNT adsorption conformers.