
Figures & data
Figure 1. Norovirus genome copy numbers and virus intra-host single nucleotide variants (iSNVs) were detected in stool samples from infected children. The genome copy number (left y-axis, circles and lines; the mean of duplicate wells) and the number of iSNVs (right y-axis, bars) were tracked in individual infection cases (episodes). The filled circles indicate qPCR and genome sequencing positive samples, while empty circles indicate qPCR positive but genome sequencing negative samples. The corresponding genotype and variant information are indicated for each episode.

Table 1. Norovirus infection episodes analysed in this study.
Figure 2. GII.4 noroviruses presented a higher number of mutational turn-over during the shedding phase of the infection. (a) The intra-host mutations were profiled into de novo (newly generated iSNVs), persisting (iSNVs observed in previous time point), or purged (iSNVs not detected) as compared to the mutational profile from the previous sample. The number of mutations was plotted on the y-axis with time intervals between corresponding samples indicated on the x-axis. *P < 0.05 and **P < 0.01 in one-way ANOVA and Sidak multiple comparisons test. (b) The dot plots summarize the number of intra-host de novo, persisting, and purged mutations by genotype, types of mutation, and ORFs. The lines and error bars indicate mean and standard deviation, respectively.
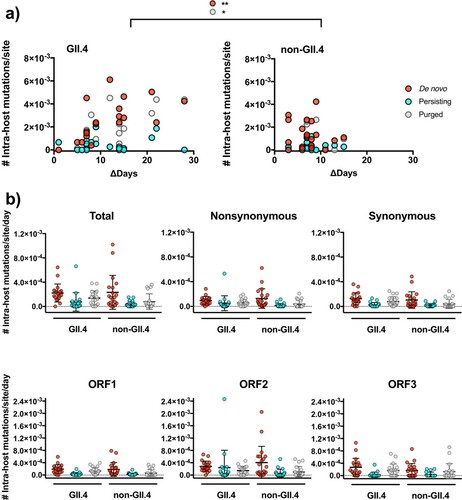
Figure 3. GII.4 noroviruses present an accumulation of nucleotide changes within the host. (a) The number of major nucleotide changes (frequency >50%) as compared to consensus genome sequence from the previous time point. The number of consensus nucleotide changes was plotted on the y-axis with time intervals between corresponding samples indicated on the x-axis. The lines in the graph indicate linear regression curves with 95% confidence intervals. (b) The dot plots summarize the number of intra-host consensus changes by genotype, types of mutation, and ORFs. The lines and error bars indicate mean and standard deviation, respectively. *P < 0.05 and **P < 0.01 in Mann–Whitney test.
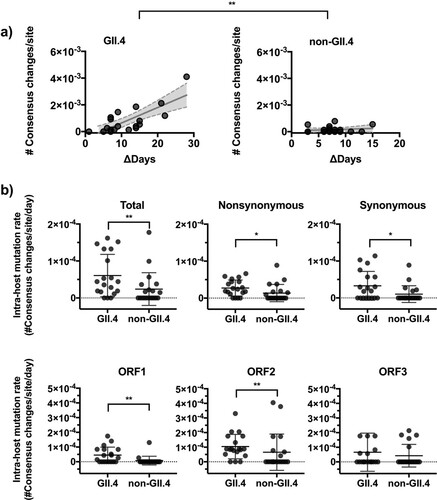
Figure 4. Nonsynonymous mutations are skewed on the P2 sub-domain on the GII.4 VP1 protein. The number of synonymous and nonsynonymous iSNVs in (a) GII.4 (7 episodes, 26 samples) and (b) GII.6 (5 episodes, 16 samples) viruses were summarized by the locations on the VP1 protein; S, P1, and P2 sub-domains.
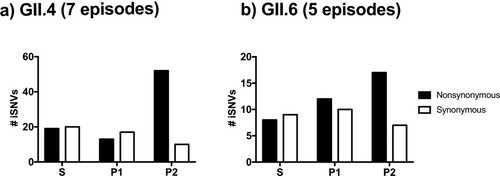
Figure 5. Major and minor intra-host mutations are differently distributed on the GII.4 VP1 protein. (a) The numbers of synonymous and nonsynonymous iSNVs were summarized by the locations on the VP1 protein: S, P1, P2 (excluding antigenic sites) sub-domains, and five major antigenic sites. Minor (frequency <50%), major (frequency ≥50%), and fixed (frequency ≥90%) iSNVs are summarized in different bar plots. (b) The intra-host amino acid mutational dynamics from residues mapping to major antigenic sites are shown for each nonsynonymous mutation. The area plots within the box indicate the amino acid changes detected in same child. The infected GII.4 variants, mutated positions, and corresponding antigenic sites are described for each area plot. The x-axis shows days since child was detected norovirus-positive. The y-axis shows the frequency of residues at each time point.
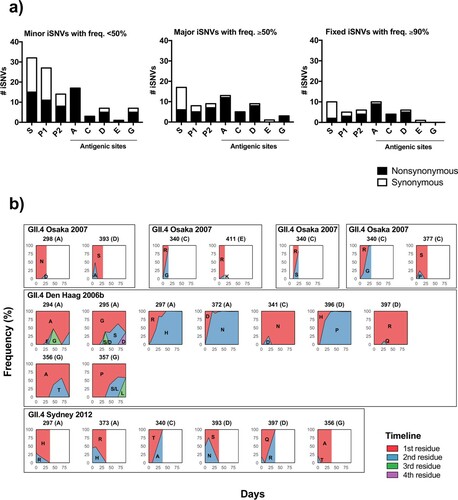
Figure 6. Intra-host mutations occurring in immunocompetent individuals are the source of GII.4 norovirus diversification on a global scale. (a) Maximum-likelihood phylogenetic tree of GII.4 noroviruses analysed in this study along with a global sequence dataset (n = 309 that were randomly selected from 1,572 complete VP1 sequences deposited in GenBank from viruses circulating during 1995–2016) indicated accumulation of amino acid changes within the host during the shedding phase; while being clustered together with viruses in a global population. Branches were colored by variants. Samples analysed in this study were denoted by circles with episode numbers; empty circles indicate viruses detected in initial samples and filled circles indicate those detected during the shedding phase of each episode. (b) Prevalence of the intra-host amino acid mutations on the P2 sub-domain in GII.4 noroviruses detected in the shedding phase was compared against observed mutations at the global level (n = 1,572 sequences). The bar plot shows the number of intra-host mutations in x-axis, colored by the prevalence of those at the end of the shedding; major intra-host mutations are indicated with black, and minor intra-host mutations with white. The y-axis indicates the prevalence of the same mutations detected using a global sequence database. From top to down: major amino acid residues detected in the same variant that infected the child; minor residues detected in the same variant infecting the child; not detected in the same variant but detected in other variants; and not detected in any GII.4 viruses. (c) The history of amino acid mutations mapping on the antigenic sites that were never detected in the same variant but dominated in the other variants was summarized in line graphs. The x-axis shows the year and the y-axis shows the ratio (%) of sequences with the specified residues in each year in the global dataset (black lines). The year of the corresponding intra-host mutation detected was indicated by a dot line in each graph.
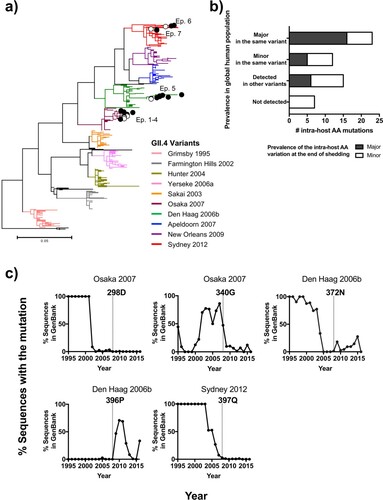
Figure 7. Intra-host mutations occurring in immunocompromised patients are not the source of GII.4 norovirus diversification on a global scale. (a) Maximum-likelihood phylogenetic tree of GII.4 noroviruses detected in immunocompromised patients along with a global sequence dataset (n = 1572) indicated disparity between viral diversity in immunocompromised patients and in a global population. Branches were colored by variants. Viruses detected from immunocompromised patients were marked based on the study [Citation18–21]; empty symbols indicate viruses detected in initial samples and filled symbols indicate those detected during the chronic infection phase. (b) Prevalence of the intra-host amino acid mutations on the P2 sub-domain in GII.4 noroviruses detected from immunocompromised patients was compared against observed mutations in the global non-immunocompromised population. The bar plot shows the number of intra-host mutations in x-axis, and the y-axis indicates the prevalence of the same mutations detected using a global sequence database. From top to down: major amino acid residues detected in the same variant that infected the immunocompromised patients; minor residues detected in the same variant infecting the immunocompromised patients; not detected in the same variant but detected in other variants; and not detected in any GII.4 viruses in the global dataset. The bar plot was colored by the variant assignment of viruses infecting immunocompromised patients; viruses assigned to known GII.4 variants were indicated with black, and those that were genetically diverged and could not be assigned to known variants were indicated with white.
![Figure 7. Intra-host mutations occurring in immunocompromised patients are not the source of GII.4 norovirus diversification on a global scale. (a) Maximum-likelihood phylogenetic tree of GII.4 noroviruses detected in immunocompromised patients along with a global sequence dataset (n = 1572) indicated disparity between viral diversity in immunocompromised patients and in a global population. Branches were colored by variants. Viruses detected from immunocompromised patients were marked based on the study [Citation18–21]; empty symbols indicate viruses detected in initial samples and filled symbols indicate those detected during the chronic infection phase. (b) Prevalence of the intra-host amino acid mutations on the P2 sub-domain in GII.4 noroviruses detected from immunocompromised patients was compared against observed mutations in the global non-immunocompromised population. The bar plot shows the number of intra-host mutations in x-axis, and the y-axis indicates the prevalence of the same mutations detected using a global sequence database. From top to down: major amino acid residues detected in the same variant that infected the immunocompromised patients; minor residues detected in the same variant infecting the immunocompromised patients; not detected in the same variant but detected in other variants; and not detected in any GII.4 viruses in the global dataset. The bar plot was colored by the variant assignment of viruses infecting immunocompromised patients; viruses assigned to known GII.4 variants were indicated with black, and those that were genetically diverged and could not be assigned to known variants were indicated with white.](/cms/asset/7ff706fc-b400-4d7c-b03e-b33462018a67/temi_a_1967706_f0007_oc.jpg)