
Figures & data
Figure 1. Definition of angular distribution function (ADF). Cmca4 is molecular, so we build a histogram based on the angles defined by the axis of a reference molecule and neighbour atoms. The histogram is weighted with the fourth power of the distance r such that the further the neighbours, the less they contribute to the distribution.
Figure 2. Upper figure, (PDOS) for I4amd and two Cmca4 structures from PBESOL harmonic lattice dynamics at a nominal pressure of 400 GPa. Lower figure, top panel shows Cmca4 PDOS extracted from 128-atom (i.e. a supercell with 8 layers of 8 molecules) , 400 GPa, 300K MD, PIMD centroid dynamics and one PIMD bead using Equation 1. The curves are normalized to the degrees of freedom of the system. bottom panel, energy distribution between the modes in MD (red), the zero-point energy obtained by quantizing the molecular dynamics PDOS (green) and the total quasi-harmonic energy (zero point and thermal) (blue).
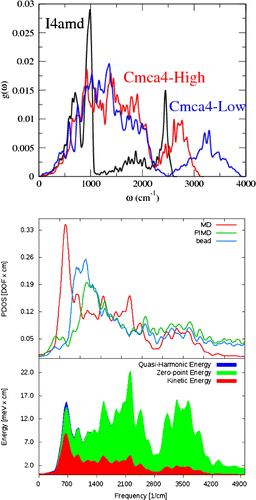
Figure 3. The radial distribution function (RDF) (top), angular distribution function (ADF) (middle) and mean square-root displacement (MSD) (bottom) for ab initio MD (red), 16-bead PIMD centroid dynamics (green) and PIMD individual bead (blue). Calculations are done for 128 atoms of Cmca4 hydrogen run at 400 GPa and 300 K, using PBE.
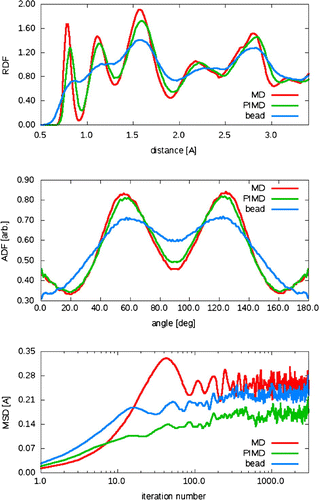
Table 1. Effect of different exchange correlation functionals on volume, Kohn–Sham enthalpy relative to I41–amd, zero-point energy. The three functionals are PBE, PBESOL and LDA; calculations are done based on static relaxations in the harmonic approximation. The relaxed fractional coordinates of the two Cmca4 structures are shown: at 300GPa the Cmca4 (high) (Pickard & Needs, Citation2007) structure transformed into the Cmca4 structure (.)
Figure 4. Results from MD and PIMD runs in NPT ensemble. Top panels and bottom right panel compare the cell volume, potential energy and total energy, respectively, as extracted from MD, PIMD (for all 128 atoms, at 300 K) and single-point DFT calculation (at 0 K, with arrows showing thermal or ZPE shifts). The total energy in DFT is the same as the potential energy and is only repeated for reference, whereas the total energy in MD is the sum of the DFT energy and vibrational energy (i.e. twice the kinetic energy in the classical harmonic limit). The last panel illustrates the zero-point plus thermal energy calculated from PIMD. The barostat is weak, and we note that all quantities vary with pressure.
