
Figures & data
Figure 1. Distribution of IAV subtypes isolated from gulls.
The numbers of viruses for which sequence data are available with the indicated subtypes isolated from gulls in North America and Eurasia are shown. The circular representation shows the total proportions when both regions are combined. Gull viruses were located by presence of M gene segment sequences in the NCBI Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html), as of September 2015.
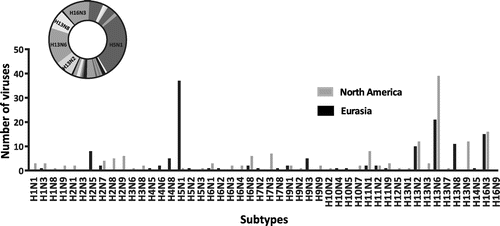
Figure 2. Time-clock Bayesian inference analysis of H13 nucleotide sequences from North America and Eurasia.
All H13 nucleotide sequences were downloaded from the NCBI Influenza Virus Resource (Bao et al., Citation2008) and aligned using MUSCLE integrated in MEGA6 (Tamura, Stecher, Peterson, Filipski, & Kumar, Citation2013). The aligned sequences were quality-trimmed, resulting in an alignment of 1133 nts (corresponding to nt positions 465–1597). This region was chosen because it allowed the inclusion of the largest number of virus sequences. We compared this tree to one constructed with a subset of viruses for which the complete segment sequences were available and the overall topology is not affected by use of this portion (not shown). The maximum credibility tree was generated from the trimmed alignment by the Bayesian inference method implemented in BEAST v1.8.0 (Drummond, Suchard, Xie, & Rambaut, Citation2012) using the SRD06 substitution model with a strict molecular clock and with the use of the GMRF Bayesian sky ride coalescent prior distribution. These parameters were chosen because they gave the best distribution of posterior probabilities. A strict molecular clock was chosen as appropriate to estimate the time of the most recent ancestors at each node because the sequences are almost all from the same host species. The branches are colored according to the host group: green, North American gulls; blue, Eurasian gulls; brown, poultry; dark blue, marine mammals; dark pink, Eurasian waterfowl; orange, North American waterfowl; purple, shorebirds and other wild birds excluding gulls, waterfowl, and poultry. The posterior probabilities for the support of the branches are given at major nodes. Photo credit: http://www.freeimages.com.
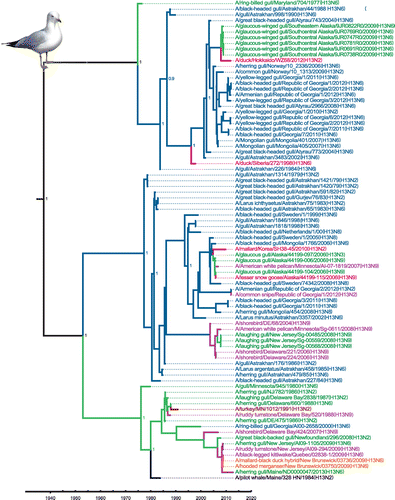
Figure 3. Time-clock Bayesian inference analysis of H16 nucleotide sequences from North America and Eurasia.
All complete H16 nucleotide sequences were downloaded from the NCBI Influenza Virus Resource (Bao et al., Citation2008) and aligned using MUSCLE integrated in MEGA6 (Tamura et al., Citation2013). The aligned sequences were quality-trimmed, resulting in an alignment of 1562 nts (corresponding to nt positions 94–1655). The maximum credibility tree was generated from the trimmed alignment by the Bayesian inference method implemented in BEAST v1.8.0 (Drummond et al., Citation2012) using the SRD06 substitution model with a strict molecular clock and with the use of the GMRF Bayesian sky ride coalescent prior distribution. The branches are colored according to the host group: green, North American gulls; blue, Eurasian gulls; brown, poultry; dark pink, Eurasian waterfowl; orange, North American waterfowl; purple, shorebirds and other wild birds excluding gulls and waterfowl. The posterior probabilities for the support of the branches are given at major nodes. Photo credit: http://www.freeimages.com.
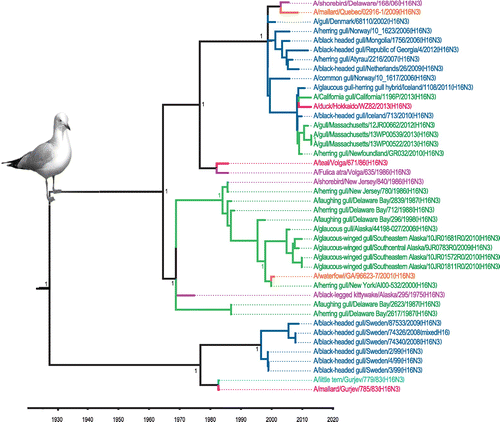
Figure 4. Putative geographic progression of gull virus genes that contributed to the genesis of the Eurasian H16N3 gull virus identified in Newfoundland, Canada.
The background map was downloaded from Mapbox Editor online tool (https://www.mapbox.com/editor).
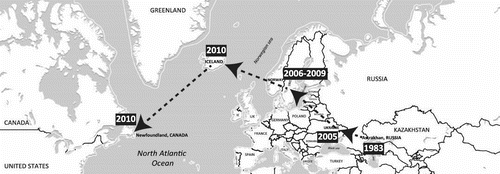
Table 1. Experimental infections of gulls with avian strains
Table 2. Experimental infections of avian and mammalian hosts with gull strains
Table 3. Gull virus binding assays on avian and human tissues (Lindskog et al., Citation2013)
Figure 5. Phylogenetic analysis of gull H5 and H7 nucleotide sequences.
Sequences of H5 and H7 genes that showed the highest similarity in BLAST (Altschul, Gish, Miller, Myers, & Lipman, Citation1990) searches to gull gene sequences were downloaded from NCBI and aligned by MUSCLE with MEGA6 (Tamura et al., Citation2013). The aligned sequences were quality-trimmed, resulting in alignments of 1,073 nts (corresponding to nt positions 172–1,242) and 1,054 nts (corresponding to nt positions 32–1,085) for the H5 and H7 gene segments, respectively. The phylogenetic tree was inferred in MEGA6 with the neighbor-joining method with 1,000 bootstrap replications. Evolutionary distances were computed using the Maximum Composite Likelihood method and are represented by the number of base substitutions per site (scale bar). The branches are colored according to the host group: green, gulls; red, waterfowl from Eurasia; orange, waterfowl from North America; pink, swine; brown, poultry; purple, shorebirds and other wild birds excluding gulls and waterfowl; blue, humans and other mammals excluding swine.

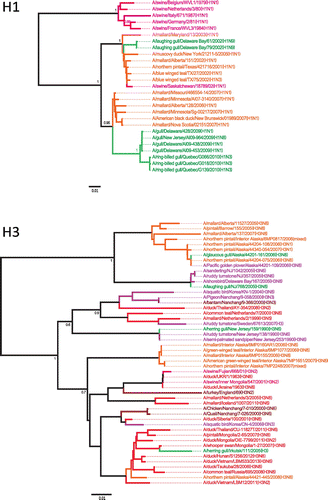
Figure 6. Phylogenetic analysis of gull H1 and H3 nucleotide sequences.
Sequences of H1 and H3 genes that showed the highest similarity in BLAST (Altschul et al., Citation1990) searches to gull gene segments were downloaded from NCBI and aligned by MUSCLE with MEGA6 (Tamura et al., Citation2013). The aligned sequences were quality-trimmed, resulting in alignments of 1,028 nts (corresponding to nt positions 10–1,037) and 641 nts (corresponding to nt positions 1,049–1,689) for the H1 and H3 gene segments, respectively. For the H3 sequences, this region was chosen because it allowed the inclusion of the largest number of virus sequences. We compared this tree to one constructed with a subset of viruses for which the complete segment sequences were available and the overall topology is not affected by use of this portion (not shown). The phylogenetic tree was inferred in MEGA6 with the neighbor-joining method with 1,000 bootstrap replications. Evolutionary distances were computed using the Maximum Composite Likelihood method and are represented by the number of base substitutions per site (scale bar). The branches are colored according to the host group: gulls, green; red, waterfowl from Eurasia; orange, waterfowl from North America; swine, pink; brown, poultry; purple, shorebirds and other wild birds excluding gulls and waterfowl.
Supplemental material