
Figures & data
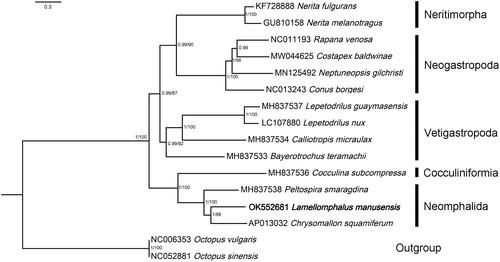
Figure 1. Relationships of Lamellomphalus manusensis to representive members of the Gastropoda. Nucleotide sequences of all protein-coding genes and ribosomal genes were individually aligned using MAFFT v. 7 (Katoh et al. Citation2019), ambiguous positions removed using GBlocks (Talavera and Castresana Citation2007), then concatenated, leading to alignments with 10,979 nucleotide positions for protein-coding genes and 1982 positions for the rRNA genes. ModelFinder (Kalyaanamoorthy et al. Citation2017) was used to select the best-fit partition model (Edge-unlinked) using BIC criterion. Maximum likelihood phylogenies were inferred using IQ-TREE (Nguyen et al. Citation2015) under the GTR + R4 + F model for 10,000 ultrafast (Minh et al. Citation2013) bootstraps, as well as the Shimodaira–Hasegawa–like approximate likelihood-ratio test (Guindon et al. Citation2010). Bayesian Inference phylogenies were inferred using MrBayes 3.2.6 (Ronquist et al. Citation2012) under partition model (2 parallel runs, 2,000,000 generations), in which the initial 25% of sampled data were discarded as burn-in. Branch support shown as maximum-likelihood bootstrap values (when ≥50)/Bayesian posterior probability (when ≥0.8).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] under the accession no. OK552681. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA798177, SRR17651057, and SAMN25049529, respectively.