Figures & data
Figure 1. Mechanism of CFTR channel gating. The CFTR channel consists of two membrane spanning domains (MSD), two nucleotide binding domains (NBD) and a single regulatory domain (RD). Following the phosphorylation of the RD by protein kinase A (PKA) and the hydrolysis of an ATP on the NBD, the CFTR channel gate moves to an open conformation and allows the passage of chloride (Cl−) ions (adapted from reference 6, Chen et al, 2006 with kind permission from S. Karger AG, Basel).
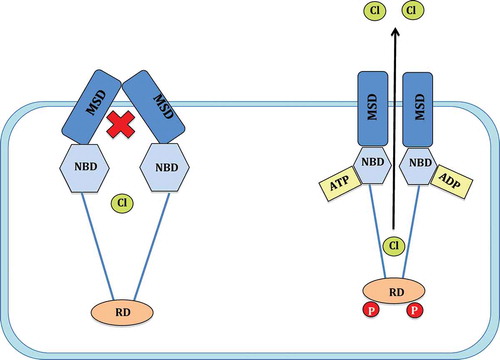
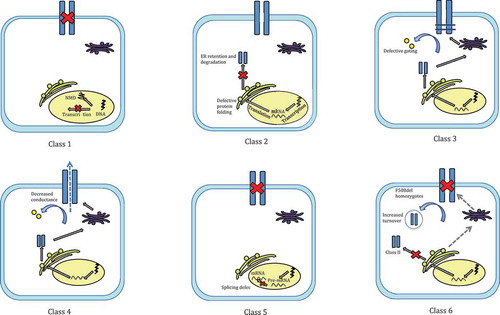
Figure 2. A schematic representation of class I to class VI mutations in cystic fibrosis. Class I mutations lead to a complete lack of production of the CFTR protein. Class II mutations result in defective handling of CFTR, typically stimulating ER retention and degradation. Class III and class IV mutations disrupt CFTR function by decreasing its gating ability and conductance respectively. Defective splicing of pre-mRNA to mRNA typically underlies class V mutations. Finally, class VI mutations increase the turn-over of CFTR at the cell membrane.