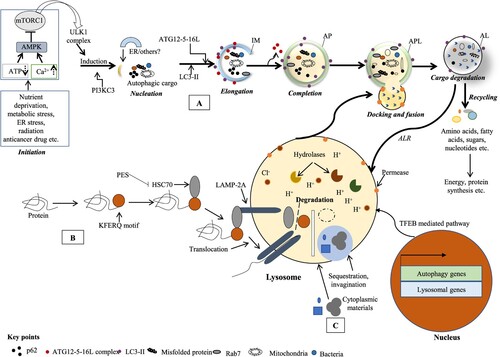
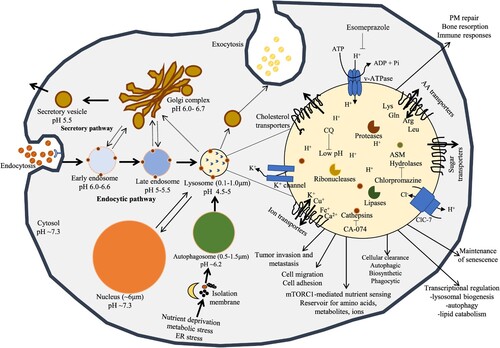
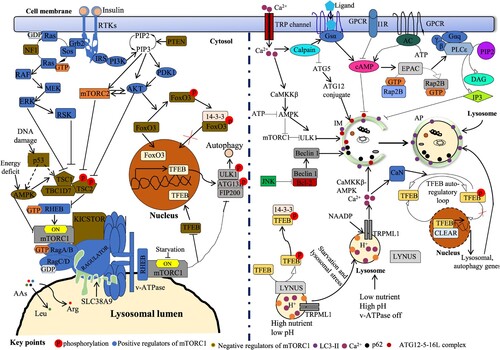
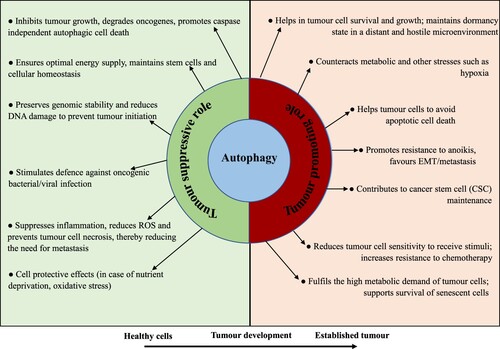
Figures & data
Table 1. Dysfunctional autophagy is associated with different human diseases.
Table 2. Selected examples of ALP inhibitors with therapeutic benefits.
Table 3. Selected examples of therapeutic benefits from ALP inhibition in animal models.
Table 4. Ongoing and completed clinical trials with ALP inhibitors.