Figures & data
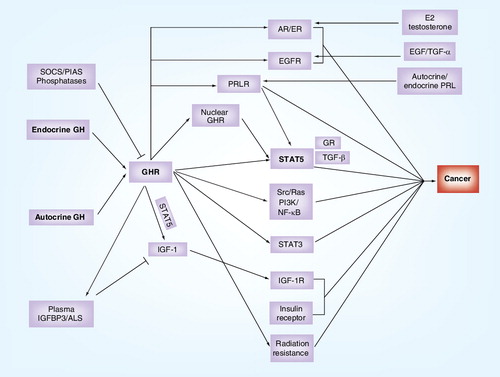
GH receptor activation by endocrine or autocrine GH is feedback regulated by SOCS, phosphatases and PIAS. Receptor activation induces transcription of IGF-1, and the receptors for prolactin, EGF, estrogen and androgen, as well as promoting insulin synthesis and secretion. While GH receptor activation results in activation of oncogenic Src, Ras/ERK, PI3-kinase and NF-κB pathways, it also activates oncogenic STATs, STAT3 and particularly STAT5. The latter complexes with glucocorticoid receptors and TGF-β, and is necessary for the transforming actions of nuclear localized GHR. Finally, IGF-1 produced as a result of GHR activation and regulated by GH-dependent IGFBP3/ALS is a key element in tumor promotion. Text in bold shows standard abbreviaitons.
ALS: Acid labile subunit; AR: Androgen receptor; EGFR: EGF receptor; ER: Estrogen receptor; GH: Growth hormone; GHR: Growth hormone receptor; GR: Glucocorticoid receptor; IGFBP3: Insulin-like growth factor-binding protein 3; PIAS: Protein inhibitors of activated signal transducer and activator of transcription; PRL: Pituitary hormone prolactin; PRLR: Pituitary hormone prolactin receptor; SOCS: Suppressor of cytokine signaling proteins; STAT: Signal transducer and activator of transcription.
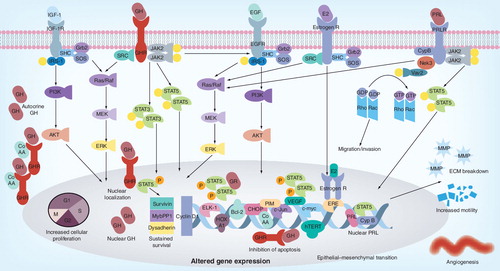
GH and PRL signaling via Jak2 mediate many of the downstream responses through phosphorylation of STAT transcription factors, MAP kinases and other kinase cascades. STAT5 and STAT3 are major contributors to the downstream signaling and upregulate key proliferative genes. In PRLR-mediated signaling the phosphorylation of Vav2 (a guanosine nucleotide exchange factor that stimulates the Rho/Rac protein family) by Nek3 allows it to enhance the activity of Rac responsible for enhanced motility and invasion. The somatotrophic effects of GH are mediated by IGF-1 and its receptor (IGF-1R) serves as a potent proliferative signaling system that stimulates cell growth and promotes cell survival. The proliferative actions of IGF-1 are mediated by the association of receptor tyrosine kinase with SHC, Grb2 and SOS that activate the Ras and MAPK cascade (Raf, Mek and Erk1/2). This cascade culminates in activation of Elk-1 and other transcription factors. The anti-apoptotic effects are mediated by the phosphorylation of IRS-1 via the PI3K pathway resulting in the activation of Akt and mTOR. A cell exhibiting upregulation of these key signaling molecules or lack of a negative feedback has increased mitogenic potential with inhibition of apoptotic pathways. GHR nuclear localization, a function of highly proliferative cells, can promote in tumorigensis via the Jak2/STAT5 pathway together with CoAA, dependent on autocrine GH. This results in dysregulation of expression of survivin, dysadherin and Mybbp1a – genes associated with tumorigenesis. PRL can also be translocated into the nucleus by binding to CypB, which enhances the STAT5 DNA-binding activity and prolactin-induced STAT5-mediated gene expression. Autocrine GH increases cell proliferation (via HoxA1, TFF3, Jak/STAT, MAPK, Cyclin D1, c-myc, hTERT), survival (via TFF3, Bcl2, CHOP, Jak/STAT) and migration/invasion/EMT (via TFF3, MMP, γ-catenin) and angiogenesis (TFF3, VEGF) by differential regulation of bracketed genes. estrogen receptor, androgen receptor and EGFR upregulated by GH signaling also contributes to tumor promotion by genomic and plasma membrane actions.
ECM: Extracellular matrix; GH: Growth hormone; GHR: Growth hormone receptor; PRL: Prolactin.