Figures & data
Reproduced with permission from Citation[153].
![Figure 1. a-1-antitrypsin structure.Reproduced with permission from Citation[153].](/cms/asset/722084d5-3b40-4f7c-b87e-18a7f3c88e02/ierx_a_11219368_f0001_b.jpg)
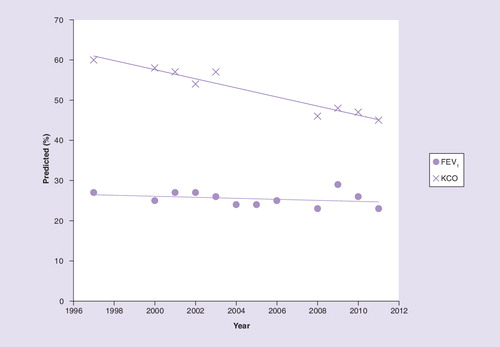
Polymerization of Z A1AT in the lung (1) leads to local ER stress (2) and establishment of a postinflammatory cascade (3) including increased neutrophil recruitment (4). Lung polymers are also chemoattractants, recruiting and localizing neutrophils further (4). Polymerization in the liver (5) results in serum and lung deficiency (6), causing unopposed neutrophil elastase activity (7) as a result of the recruited neutrophils (4), which in turn can activate other classes of enzymes (8) in addition to other uninhibited serine proteinases. The net result is proteolytic degradation of lung tissue (9), leading to emphysema. In addition, neutrophil elastase-derived peptide (10) and chemokines (11) can amplify the neutrophilic load and accelerate parenchymal damage through further release of proteinases.
A1AT: α-1-antitrypsin; ER: Endoplasmic reticulum; LTB4: Leukotriene B4;
MMP: Matrix metalloproteinase; PR3: Proteinase 3.
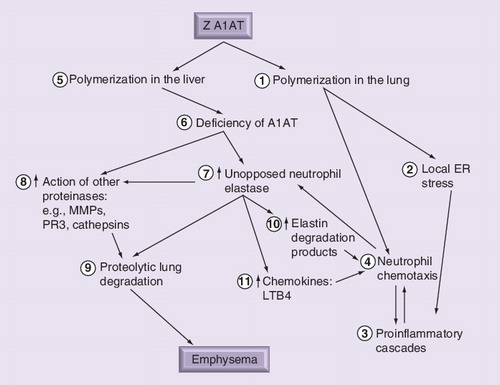
Local deficiency of A1AT leads to excess unopposed neutrophil elastase activity (1). Elastase binds to macrophages stimulating release of leukotriene-B4 (2), a potent neutrophil chemotactic mediator. This promotes neutrophil recruitment (3), causing further release of neutrophil elastase and tissue damage (4). In addition, mutant A1AT polymers colocalize with neutrophils, retaining them in the interstitium and promoting further connective tissue degradation (5).
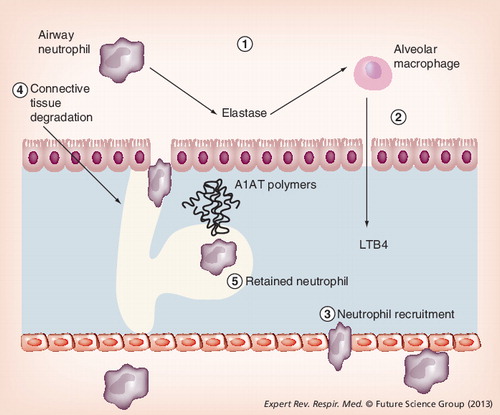
FEV1: Forced expiratory volume in 1 s; KCO: Carbon monoxide transfer coefficient.