
Figures & data
Figure 1 Mechanism of action of agents used in the treatment of BC. In general, conventional therapeutic treatments for the management of BC patients include endocrine therapy, targeted therapy, and chemotherapy. Standard therapy for the treatment of ER+ BC is typically based on the use of endocrine therapy (TAM, FUL, and AIs). Additional treatments include CDK4/6 inhibitors, PI3K inhibitors, and drug repurposing. Treatments approved for HER2+ BC include humanized antibodies (trastuzumab, pertuzumab, and 19H6-Hu), PI3K inhibitors, antibody-drug conjugates (trastuzumab emtansine and trastuzumab deruxtecan), TKI inhibitors, mTOR inhibitors, nanotechnological approaches (nanocarriers), and CRISPR. Treatments for TNBC include chemotherapy and immunotherapy (bevacizumab), among others.
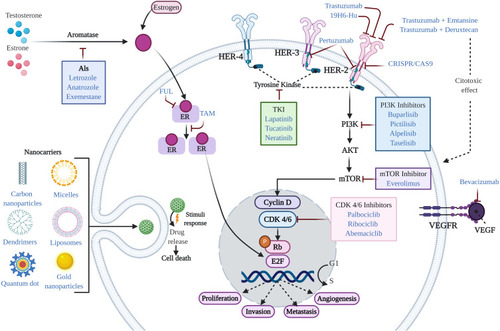
Figure 2 Mechanisms of resistance to endocrine therapy with tamoxifen (TAM) in breast cancer. (A) Resistance in BC is characterized by the deficiency of CYP2D6, which reduces the metabolism of 4OH-TAM. (B) Acquired resistance to TAM involves alterations in translation signals by increasing the expression and activity of tyrosine kinase receptor family proteins, such as HER2, EGFR, IGFR and GPR30. These events lead to aberrant activation of cAMP/PKA, MAPK/ERK and PI3K/AKT signaling pathways. Activation of these kinase pathways can result in phosphorylation of ER and its co-activators such as A1B1, MED1, or CARM1, leading to the activation of proliferation and inhibition of apoptosis. (C) Deregulation of ER increases not only HER2-mediated signaling, but the activation of transcription factors such as Sp1, AP-1 and NFκB, promoting oncogene transcription. Likewise, factors such as FOXA1 and PBX1 can recruit ER to specific genomic sites. (D) Other mechanisms that can lead to resistance to TAM involve the activation of alternate signaling pathways; for example, mutations in the tumor suppressor protein, PTEN, has been shown to increase phosphorylation of PI3K/AKT in ER+ tumors, leading to therapeutic resistance. Additionally, aberrant phosphorylation of proteins that modify histones, such as KDM6B and EZH2, can lead to resistance to endocrine therapy from the reactivation of genes.
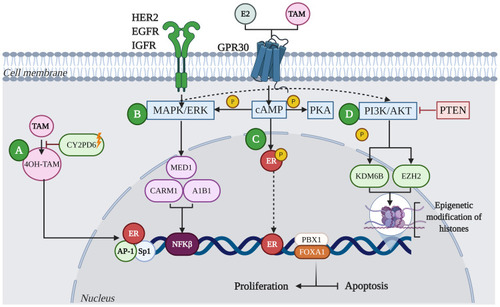
Table 1 Some Repurposed Drugs for Breast Cancer Treatment
Figure 3 Mechanisms leading to the generation of resistance to chemotherapy. (A) Effusion pumps: Membrane glycoproteins act as pumps extracting the drug efflux of intracellular space, thus reducing drug concentration and efficacy. (B) Receptor affinity: The number and affinity of receptors present on the membrane determine the effectiveness of the drug, allowing it to be paired or not. (C) The enzyme system involved in the deactivation of anti-cancer drugs, and therefore involved in the process of drug resistance, includes multi-drug resistance gene (MDR1) and glutathione S transferase Pi (GSTP1). While GSTP1 decreases the concentration and the effective life of drugs, resulting in drug inefficiency, MDR1 gene is significantly overexpressed in multidrug resistance phenotype. (D) Epigenetic mechanisms: DNA methylation and histone modifications [acetylation (Ac) or methylation (Me)] lead to the silencing of tumor suppressor genes and/or overexpression of oncogenes. (E) Tumor microenvironment: the tumor microenvironment creates the optimal niche for the development of cancer cells. In conditions such as hypoxia, cancer cells interact with stromal and immune system cells through exosomes from which they acquire nutrients to use other metabolic pathways, leading to the overexpression of the MRP and deactivating and extracting the drug from inside the cell. Additionally, hypoxia is a trigger for tumor resistance to chemotherapy, since it leads to both decreased DNA topoisomerase II alpha (TOP2A) expression and upregulation of MRP. (F) Angiogenesis: The TWIST1 transcription factor can recognize the E-box gene sequence on the promoter of E-cadherin and depress its transcription, thereby leading to decreased cell adhesion and promoting angiogenesis.
![Figure 3 Mechanisms leading to the generation of resistance to chemotherapy. (A) Effusion pumps: Membrane glycoproteins act as pumps extracting the drug efflux of intracellular space, thus reducing drug concentration and efficacy. (B) Receptor affinity: The number and affinity of receptors present on the membrane determine the effectiveness of the drug, allowing it to be paired or not. (C) The enzyme system involved in the deactivation of anti-cancer drugs, and therefore involved in the process of drug resistance, includes multi-drug resistance gene (MDR1) and glutathione S transferase Pi (GSTP1). While GSTP1 decreases the concentration and the effective life of drugs, resulting in drug inefficiency, MDR1 gene is significantly overexpressed in multidrug resistance phenotype. (D) Epigenetic mechanisms: DNA methylation and histone modifications [acetylation (Ac) or methylation (Me)] lead to the silencing of tumor suppressor genes and/or overexpression of oncogenes. (E) Tumor microenvironment: the tumor microenvironment creates the optimal niche for the development of cancer cells. In conditions such as hypoxia, cancer cells interact with stromal and immune system cells through exosomes from which they acquire nutrients to use other metabolic pathways, leading to the overexpression of the MRP and deactivating and extracting the drug from inside the cell. Additionally, hypoxia is a trigger for tumor resistance to chemotherapy, since it leads to both decreased DNA topoisomerase II alpha (TOP2A) expression and upregulation of MRP. (F) Angiogenesis: The TWIST1 transcription factor can recognize the E-box gene sequence on the promoter of E-cadherin and depress its transcription, thereby leading to decreased cell adhesion and promoting angiogenesis.](/cms/asset/b2a146f8-e181-4e78-a149-8427900c8ccd/dbct_a_33204149_f0003_c.jpg)
Table 2 miRNAs in the Response to Breast Cancer Therapy