Figures & data
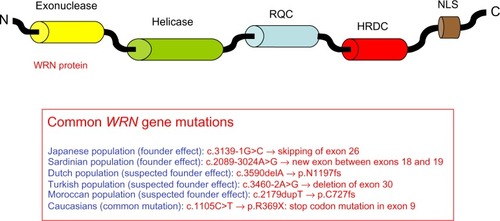
Figure 1 Representation of prelamin A processing and summary of LMNA and ZMPSTE24 mutations leading to classical and atypical progeria.
Notes: Prelamin A requires maturation to become mature lamin A. First, the C-terminal end of prelamin A contains a CaaX motif, which is modified by farnesylation of the cysteine residue (1), mediated by a FTASE protein, and is followed by the cleavage of the (2)
–aaX terminal residues that can be performed either by RCE1 or by ZMPSTE24. (3) After the cleavage, the cysteine residue is carboxymethylated by ICMT. The last step removes the C-terminal 15 residues through cleavage by (4) ZMPSTE24, yielding mature lamin A. In HGPS cells bearing the common
LMNA p.G608G mutation, the activation of the cryptic splice site results in the deletion of a 50aa region from prelamin A (indicated in dark blue in the figure), which contains the ZMPSTE24 cleavage site (indicated in the figure). As a result, the p.G608G mutation leads to the production and the accumulation of a smaller prelamin A protein, which cannot undergo complete maturation, termed progerin. A diagnostic test is available for the common p.G608G mutation, as well as for the less common LMNA splicing mutations, namely p.G608S, p.v607v, or IVS11+1G>A, all resulting in the activation of the cryptic splice site. Several other
LMNA mutations have been reported in a small number of patients and these lead to a spectrum of progeroid phenotypes ranging from mild–moderate to very aggressive forms, and are referred to as atypical progeroid syndromes. Those mutations can be either dominant (in red ink) or recessive (in green ink) and can alter residues throughout the protein structure with no clear clustering in a single region of lamin A. The term (ch) indicates a compound heterozygous mutation
(in orange ink). Underlined LMNA mutations are those observed in atypical WS cases. Recessive ZMPSTE24 mutations also disrupt prelamin A processing and are associated with three distinct, but related, human diseases that share features of premature aging, with a gradation of severity: mandibuloacral dysplasia, atypical HGPS, and restrictive dermopathy. Concerning ZMPSTE24 mutations, the term (ch) indicates a compound heterozygous mutation (in orange ink), otherwise the mutation was found in homozygosis in the patient (in green ink).
Abbreviations: HGPS, Hutchinson–Gilford Progeria Syndrome; ZMPSTE24, zinc metalloprotease related to Ste24p; FTASE, farnesyltransferase; RCE1, Ras-converting enzyme 1; ICMT, isoprenylcysteine carboxyl methyltransferase; WS, Werner syndrome.
Abbreviations: HGPS, Hutchinson–Gilford Progeria Syndrome; ZMPSTE24, zinc metalloprotease related to Ste24p; FTASE, farnesyltransferase; RCE1, Ras-converting enzyme 1; ICMT, isoprenylcysteine carboxyl methyltransferase; WS, Werner syndrome.
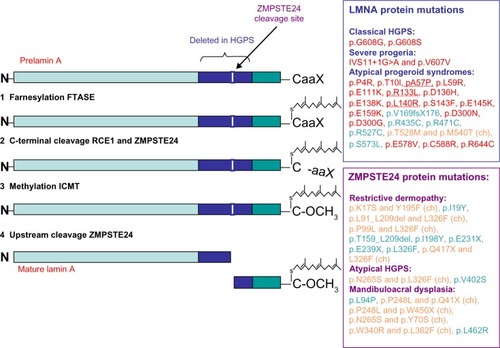
Figure 2 Representation of the WRN protein and summary of the most common WRN mutations leading to Werner syndrome.
Notes: The diagram illustrates the functional domains of the WRN protein: exonuclease, helicase, RQC, HRDC, and the NLS. Over 70 insertion/deletion mutations, missense and nonsense mutations, as well as splice mutations have been found in the different domains of the WRN protein. Genetic screening is performed as routine diagnostic tests, and the figure shows the most frequent mutations observed according to ethnic group.
Abbreviations: RQC, RecQ C-terminal domain; HRDC, helicase and RNase D C-terminal domain; NLS, nuclear localization signal; N, amino terminal; C, carboxyl terminal.
Abbreviations: RQC, RecQ C-terminal domain; HRDC, helicase and RNase D C-terminal domain; NLS, nuclear localization signal; N, amino terminal; C, carboxyl terminal.