Figures & data
Figure 1 Oxidative stress-induced DNA damage may stimulate the assembly of DNAPKsome formation and activation of Mps1, which phosphorylates c-Abl at Threonine 735 (T735), promoting its cytoplasmic translocation. c-Abl phosphorylates the Tyrosine 56 (Y56) in the PIM domain of OTULIN, disrupting its association with LUBAC. The released LUBAC interacts with SPATA2 and is involved in the TNF-R1-mediated signaling pathway, which could also be activated by oxidative stress. Oxidative stress may also directly activate ATM, which phosphorylate the scaffolding subunit of PP2A, PR65, at Serine 401 (S401). This phosphorylation disassembles the holoenzyme, causing the translocation of phosphorylated PR65 to the cytoplasm and retention of the catalytic subunit, PP2A(C), in the nucleus. P2A(C) interacts with and dephosphorylates γ-H2AX. In case of severe DNA damage, nuclear ATM activation may lead to the phosphorylation of c-Abl at S465, which may interfere with DNAPKsome formation and facilitate apoptotic processes.
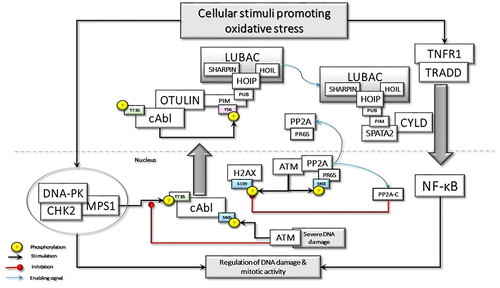
Figure 2 TRADD in the TNFR1sc recruits the TRAF2, c-IAPs, and RIPK1 through dead domain (DD) interactions. Membrane-associated TRAF2 comes close to sphingosine kinase (SpK1) and binds with it. SpK1 may activate the E3 ligase function of TRAF2 through its product sphingosine-1-phosphate (SP1). Subsequently, K63-linked autoubiquitination of TRAF2 promotes the recruitment of downstream effector kinases, such as MEKK1 and TANK. TRAF2-TANK interaction stimulates the activation of IKKϵ and TBK1. IKKϵ phosphorylates TRAF2 at Ser11, which may promote the interaction of ubiquitinated TRAF2 with the UBA domain of c-IAPs. Then, the stabilized TRAF2-c-IAP interaction drives the K63-linked ubiquitination of IKKϵ, RIPK1, and TRADD for the recruitment of LUBAC.
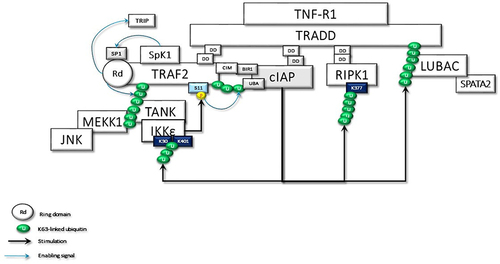
Figure 3 The activated and ubiquitinated IKKϵ may interact with the second CAP-Gly UBD domain of TRAF2-associated CYLD. This interaction enables IKKϵ to phosphorylate Ser418, which may induce the dimerization of CYLD. Then, the dimerized CYLD is associated with the SPATA2-LUBAC complex. Thus, IKKϵ phosphorylation promotes the LUBAC-associated transportation of CYLD to TNF-Rsc. Furthermore, the dimerization of CYLD also may induce the interaction with MIB2, NEMO, and ITCH. In addition to augmenting protein interactions through scaffolding activity, CYLD protects the MIB2 and LUBAC degradation caused by autoubiquitination.
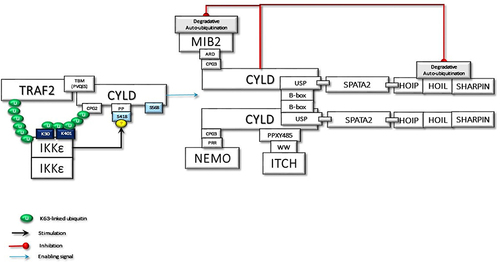
Figure 4 The CYLD-SPATA2-LUBAC association enhances the linear ubiquitination function of LUBAC. Activated LUBAC, associated with K63 linked ubiquitin chains on RIPK1, adds Met1-linked ubiquitin chains on K63-linked ubiquitin chains. K63/Met1-hybrid ubiquitin chains on RIPK1 recruit NEMO for the activation of canonical IKKs. RIPK1 in the TNF-R1sc interacts with the MZM motif of MIB2 through its linker region. Thus, activated MIB2 autoubiquitinates itself with K63-linked chains, which facilitates its interaction with the third CAP-Gly domain of CYLD by its ankyrin repeat domain (ARD). MIB2 also interacts by its zinc finger (ZF) motif to the caspase domain (CasD) of c-FLIPL and then decorates it with K63-linked ubiquitin chains. Furthermore, CYLD-associated LUBAC conjugates M1-linked ubiquitination chains on c-FLIPL. Thus, the hybrid K63/Met1 ubiquitin chains stabilize c-FLIPL to inhibit the caspase-8 activity. In addition to the scaffolding activities, K63-linked and Met1-linked poly-ubiquitin chains on RIPK1 block its kinase activity. K63-linked ubiquitin chains on MIB2 interact with TAB proteins for the recruitment and the activating phosphorylation of TAK1, which is also associated with CYLD-bound ITCH. Following TAB-driven interaction with TAK1, MIB2 may ubiquitinate the K562 residue, which may promote autophosphorylation of TAK1 at T187 and subsequent phosphorylation at T189 (significance was denoted by an asterisk (*) and it has been moved to a free area and detailed in order not to confuse the main shape further in the figure). Besides K63-linked poly-ubiquitin chains on RIPK1, K63-linked chains on MIB2 may also provide support for the activation of CYLD-associated NEMO by providing an additional scaffold. PLK1 interaction with CYLD and RIPK1 may alter its mitotic activity as detailed in the text.
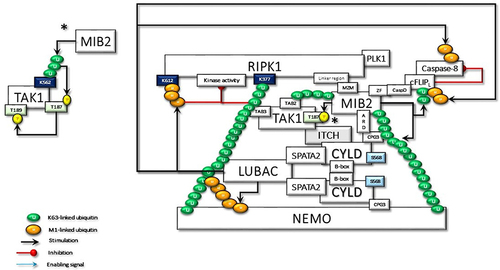
Figure 5 The activated TAK1 phosphorylates IKKα and IKKβ complexed with NEMO, which is required for the NF-κB transcriptional activity. In the absence of regulation to terminate the signaling pathway, NFK stimulates the expression of many genes, some of which promote persistent activity. IKKβ also phosphorylates Ser568 of CYLD to stimulate its DUB activity. The activated CYLD deconjugates K63-linked ubiquitin chains that trigger a cascade of reactions to terminate signal transduction.
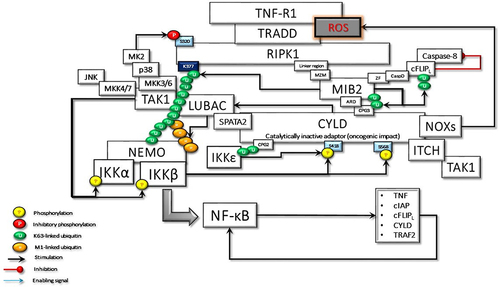
Figure 6 The activated CYLD behaves as a tumor suppressor and deubiquitinates the K63-linked ubiquitin chains on TRAF2, RIPK1, and MIB2. Loss of K63-linked chains reason for the stimulation of the K48-linked degradative ubiquitination of TAK1 and c-FLIPL by ITCH, and CYLD by MIB2, thus terminating CYLD-mediated signaling events.
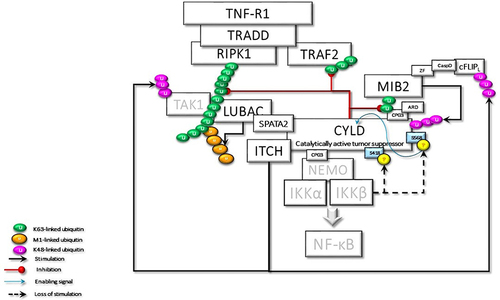
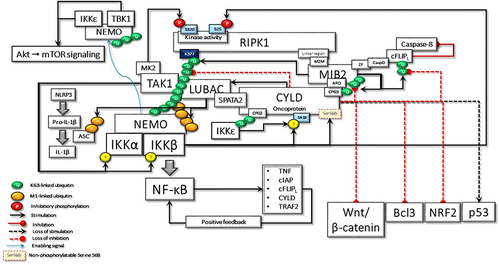
Figure 7 In the case of hypothetical loss of IKKβ-mediated Ser568 phosphorylation, CYLD behaves as an oncoprotein, keeping its scaffolding activity. Here, some of the important cyclical activities that can lead to cell transformation due to the loss of CYLD DUB activity are summarized: i) uncontrolled NF-κB activation due to positive feedback regulation; ii) RIPK1 kinase activity-dependent and -independent resistance to apoptosis; iii) IKKϵ- and TBK1-stimulated increased Akt and mTOR signaling; iv) LUBAC-induced increased secretion of IL-1β; v) Bcl3-driven increased transcriptional activity; vi) NRF2-driven increased transcriptional activity; vii) increased Wnt-β-catenin signaling; viii) loss of p53-mediated transcriptional activity; ix) MIB2-c-FLIPL-mediated caspase 8 inhibition; x) Increased Akt-mTOR signaling; xi) increased IL-1β secretion.