Figures & data
Figure 1 Epithelial differences between normal and diseased small airways. Ciliated, cuboidal epithelial cells constitute the majority of a normal bronchiolar epithelium. In a diseased SAE, however, these cell types undergo metaplasia and become a more squamous phenotype. Secretory goblet cells are found in small numbers within a normal SAE, but under diseased conditions, increase in prevalence. By contrast, the number of secretory club cells are reduced in a diseased SAE, compared to normal, with a subsequent reduction in the secretion of the protective protein, CC-16. Their proposed progenitor cell, the basal cell, is however present in greater abundance in a diseased epithelium, compared to normal.
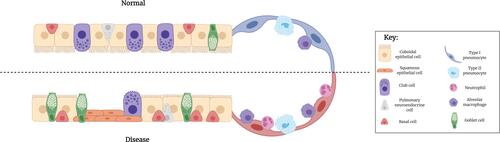
Figure 2 Activation of transmembrane RAGE in COPD. (A) The soluble isoform of receptor for advanced glycation endproducts (sRAGE) can arise from (i) alternative mRNA splicing or (ii) through ADAM10-mediated proteolytic cleavage of the transmembrane isoform. (B) Under normal physiological conditions, sRAGE performs as a decoy receptor; ligands of RAGE preferentially bind to the soluble isoform, creating a complex that is then degraded. Studies in the context of COPD have shown an enhanced expression of transmembrane RAGE and its ligands, as well as a decreased concentration of sRAGE. Consequently, RAGE ligands are able to bind to their cognate receptor, initiating a downstream pathway that eventuates in an inflammatory response. Dia-1, a Rho effector protein, is bound to the C-terminal of RAGE and is responsible for activation of a Rho-like small GTPase: Rac1. NADPH oxidases (NOXes) signal through Rac1 to generate downstream reactive oxygen species (ROS). ROS are an intracellular signaling intermediate, which increase tyrosine kinase activity and initiate the PI3K/Akt pathway. Activation of Akt promotes the nuclear translocation of NFkB and subsequent transcription of several genes, including pro-inflammatory cytokines TNF-ɑ and interleukin- (IL-) 1, 2, 6, 8 and 12. Alternative pathways of NFkB initiation are achieved through p21/Ras-ERK1/2 or p38 activation. A positive feedback loop is initiated by the NFkB transcription of RAGE.
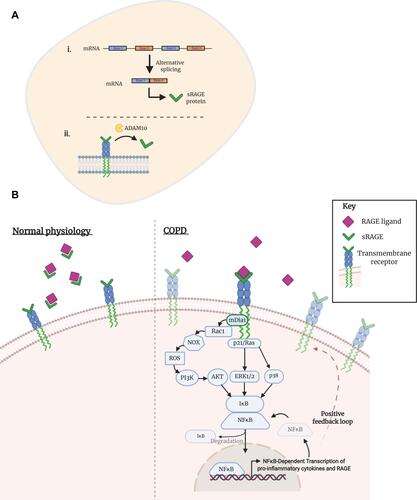
Table 1 Summary of Evidence Surrounding the Role of MMP-9 and -12 in COPD and Emphysema
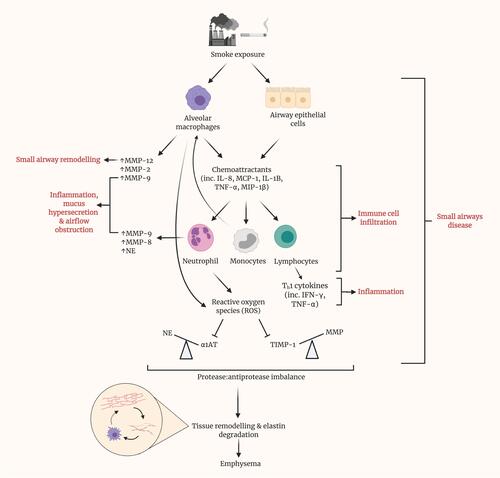
Figure 3 Prolonged smoke exposure initiates various pathways, resulting in disease of the small airways and eventuating in the onset of emphysema. Upon exposure to smoke, both alveolar macrophages and epithelial cells within the small airways release various chemoattractants, including interleukin (IL)-8, monocyte chemoattractant protein (MCP)-1, IL-1B, tumor necrosis factor (TNF)-α and macrophage inflammatory protein (MIP)-1β. These, in turn, recruit neutrophils, monocytes and lymphocytes and promote their infiltration of the small airways. Alongside alveolar macrophages, neutrophils are a source of matrix metalloproteinases (MMPs) within the lungs. MMPs have been shown to contribute to several of the phenotypic changes in SAD, namely remodeling, inflammation, mucus hypersecretion and airflow obstruction. Recruited monocytes differentiate into macrophages, contributing almost to a positive feedback loop. Lymphocytes represent a major source of cytokines, particularly Th1 cytokines such as IFN-γ, which contribute greatly to local inflammation. Reactive oxygen species (ROS) generation by neutrophils inhibits the action of antiprotease, alpha-1 antitrypsin (AAT), thus causing an imbalance with its cognate protease, neutrophil elastase. Similarly, the antiprotease activity of TIMPs is hindered by ROS. By result, a large imbalance is observed, in favor of protease activity. Cumulatively, these prior pathways culminate in tissue remodeling and elastin degradation within the alveoli, leading to emphysema. Elastin fibers, as liberated by elastin degradation, activate alveolar macrophages.