
Figures & data
Figure 1 Schematic representation of breach in host innate immunity due to different bacterial infections and development of bacteremia and sepsis.
Notes: Once the bacteremia is developed in the host and the initial control of systemic infection fails, it leads to increased activation of the innate immune response systemically. This exaggerated innate immune response causes MOD or MODS. The development of MODS leads to the ultimate death of the patients.
Abbreviations: ARDS, acute respiratory distress syndrome; DIC, disseminated intravascular coagulation; IBD, inflammatory bowel disease; MOD, multiorgan damage; MODS, multiorgan dysfunction syndrome.
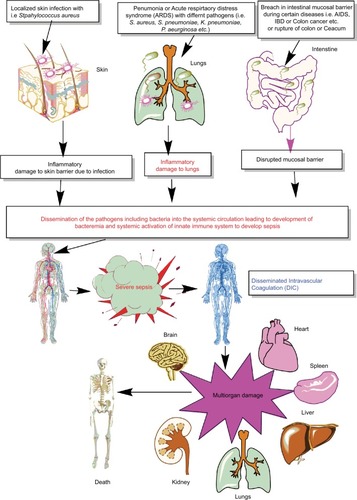
Figure 2 Schematic representation of canonical and non-canonical inflammasome signaling pathways.
Notes: The figure describes both the canonical and non-canonical signaling pathways involved in the activation of different inflammasome components including NLRP1, NLRP3, NLRC4, and AIM2. The canonical inflammasome signaling pathway causes activation of procaspase 1 into mature CASP1 that cleaves immature proIL-1β and proIL-18 into mature IL-1β and IL-18 involved in the generation of inflammatory immune response. The intracellular LPS and type-1 IFNs cause induction of non-canonical signaling in macrophages and DCs via the induction of procaspase 11 that further activates NLRP3 inflammasome by inducing a decrease in cytosolic K+ through different mechanisms described in the text. Additionally, CASP11 cleaves GSDMD into N- and C-terminal components that, upon their cytosolic accumulation, form GSDMD oligomer causing cell death, while GSDMD-NT causes pyroptosis, but it does not cause the death of adjacent cells upon its extracellular release. Detailed mechanism of inflammasome signaling pathway is mentioned elsewhere in the text.
Abbreviations: DAMP, death/damage-associated molecular pattern; DCs, dendritic cells; GSDMD, gasdermin D; IFN, interferon; LPS, lipopolysaccharide; PAMP, pathogen-associated molecular pattern.
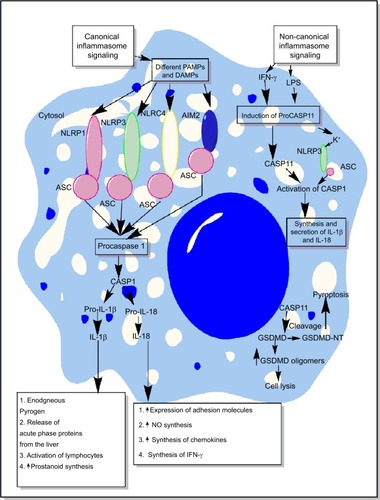
Figure 3 Schematic representation of the role of inflammasomes and their regulation via different molecules or factors elevated during sepsis.
Notes: (A) This part figure shows the induction of NLRP3 inflammasomes during sepsis in macrophages, causing their death, release of high-mobility group box 1 protein (HMGB1), microparticles (consisting of IL-1β, NLRP3, ASC, and CASP1) binding to endothelial cells and stimulating the induction of higher expression of adhesion molecules (ICAM-1 and VCAM-1), causing profound infiltration of neutrophils at the site of infection, which leads to increased tissue or organ damage and death of endothelial cells, in turn leading to increased circulating levels of ICAM-1 and VCAM-1, MOF, and finally death of the septic patient. MCC950, an NLRP3 inflammasome inhibitor, caused inhibition of increased expression of ICAM-1 and VCAM-1 on the endothelial cells and prevented MOF. (B) This part figure shows increased circulating levels of ATP and its metabolite called adenosine act via different receptors called P2X7 and A2ARs expressed on the neutrophils, DCs, and macrophages. The binding then activates the NLRP3 inflammasomes and CASP1 to release IL-1β and IL-18. These inflammatory cytokines cause increased neutrophil infiltration and tissue/organ damage during sepsis. The P2X7 receptor antagonist is shown to inhibit the activation of NLRP3 inflammasome and, thus, the inflammatory tissue damage. Furthermore, activation of NLRP3 inflammasomes decreases the process of autophagy that contains the inflammation, as restoring autophagy has been used to target sepsis. See text for full details.
Abbreviations: ATP, adenosine 5′-triphosphate; DC, dendritic cell; ICAM, intracellular adhesion molecule; MOF, multiorgan failure; VCAM, vascular cell adhesion molecule.
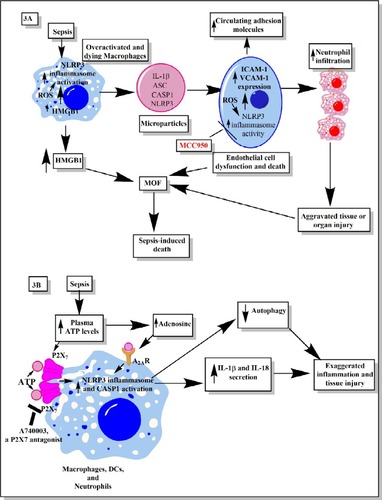
Figure 4 Role of complement in the activation of inflammasomes in innate immune cells during sepsis.
Notes: The complement (C3b)-opsonized bacteria phagocytosed via MAC cause the binding of MAC to the macrophage membrane, which leads to the initiation of bystander damage via the upregulation and activation of NLRP3 inflammasome. The activation of NLRP3 causes activation of CASP1, which leads to the maturation and release of proinflammatory cytokines, IL-1β and IL-18. On the other hand, the sublethal form of the MAC called sublethal MAC also induces the activation of NLRP3 inflammasome and the release of IL-1β and IL-18 via increasing the intracellular Ca2+ which enters into the mitochondrial matrix. The increase of Ca2+ in the mitochondrial matrix leads to increased mtROS generation and alters the mitochondrial membrane potential, causing mitochondrial damage and the release of mitochondrial DNA into the cytosol. All these factors activate NLRP3 inflammasome. Additionally, binding of C3a to its cognate receptor C3aR also activates NLRP3 inflammasome via activating the ERK1/ERK2 pathway which causes the efflux of ATP that activates NLRP3 inflammasome via binding to P2X7. The ATP that gets converted into adenosine via the enzymatic action of CD39 and CD73 also activates NLRP3 inflammasome by binding to A2AR.
Abbreviations: A2AR, A2A receptor; ATP, adenosine triphosphate; MAC, membrane attack complex; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species.
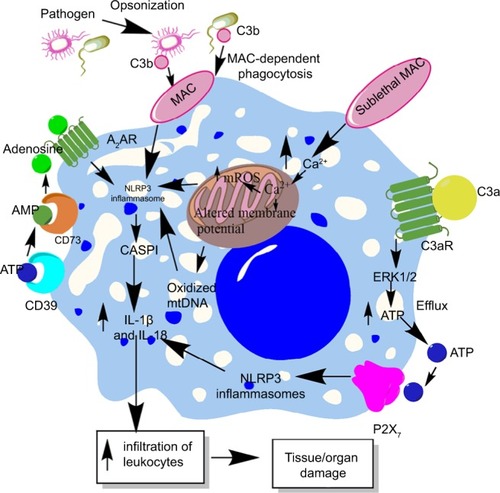
Figure 5 H2S production during sepsis and its impact on inflammasomes.
Notes: H2S is produced by the enzymatic action of CSE, CBS, CAT, and MST on the amino acids l-cysteine and homocysteine (for details, see wangCitation271) At a lower level, H2S inhibits NLRP3 inflammasome activation and the release of IL-1β and IL-18 via inhibiting the generation of mtROS and CHOP activity required for the activation of CASP1 and CASP11. However, at its higher levels observed during severe sepsis or septic shock, H2S exerts proinflammatory action via enhancing the levels of IL-1β and IL-18 through activating the CASP1, AIM2, and NLRP1b inflammasomes.
Abbreviation: mtROS, mitochondrial reactive oxygen species.
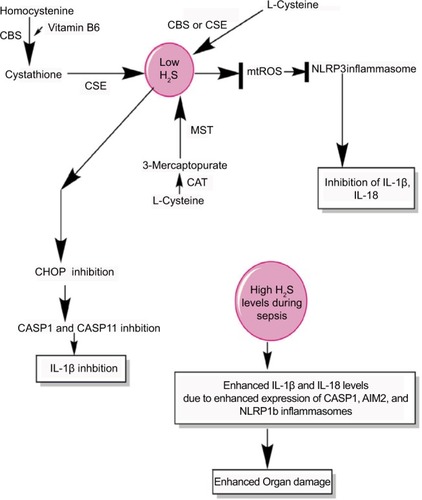
Figure 6 Schematic representation of the relationship between prostanoids, eicosanoids, sepsis, and inflammasomes.
Notes: Overactivation of inflammasomes is seen in sepsis, which also exhibits overproduction of prostanoids and eicosanoids. These prostanoids and eicosanoids exert their impact on vascular tone and vascular contractility and induce vascular leakage. Additionally, at the immune cell level, a prostanoid called PGE2 causes induction of PKA via acting through EP4 receptors expressed on the macrophages, which directly inhibits NLRP3 inflammasome by binding to Ser295. Also, PGE2 activates RIPK2 via EP3, which activates MAP kinases and NF-κB causing activation of NLRC4 inflammasomes. The activation of NLRC4 inflammasome increases the systemic levels of IL-18 and IL-1β cytokines, while PGD2, via acting through DP1, induces the production of PYDC3 protein that directly inhibits NLRP3 inflammasome overactivation. See text for detail.
Abbreviations: 5,14-HEDGE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine; 20-HETE, 20-hydroxyeicosatetraenoic acid; DP1, D-prostanoid receptor 1; PG, prostaglandin.
![Figure 6 Schematic representation of the relationship between prostanoids, eicosanoids, sepsis, and inflammasomes.Notes: Overactivation of inflammasomes is seen in sepsis, which also exhibits overproduction of prostanoids and eicosanoids. These prostanoids and eicosanoids exert their impact on vascular tone and vascular contractility and induce vascular leakage. Additionally, at the immune cell level, a prostanoid called PGE2 causes induction of PKA via acting through EP4 receptors expressed on the macrophages, which directly inhibits NLRP3 inflammasome by binding to Ser295. Also, PGE2 activates RIPK2 via EP3, which activates MAP kinases and NF-κB causing activation of NLRC4 inflammasomes. The activation of NLRC4 inflammasome increases the systemic levels of IL-18 and IL-1β cytokines, while PGD2, via acting through DP1, induces the production of PYDC3 protein that directly inhibits NLRP3 inflammasome overactivation. See text for detail.Abbreviations: 5,14-HEDGE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine; 20-HETE, 20-hydroxyeicosatetraenoic acid; DP1, D-prostanoid receptor 1; PG, prostaglandin.](/cms/asset/ee925bbc-f3a6-4579-91ad-ae7d5c1ec8d4/djir_a_178084_f0006_c.jpg)