
Figures & data
Figure 1 Signaling pathway of apoptosis. Intrinsic apoptotic pathway: Intracellular damage signals make mitochondrial outer membrane permeabilization (MOMP) release cytochrome C and the second mitochondria-derived activator of caspases (SMAC) into the cytoplasm. Cytochrome C integrates with apoptotic protease activating factor-1 (APAF-1) and pro-caspase - 9 to establish apoptosomes. In the apoptosomes, caspase-9 self-activates and reactivates the apoptotic “executioner” function of caspase-3/7. At the same time, SMAC impedes the function of inhibitors of apoptosis proteins (IAPs) and fosters apoptosis. BH3-only proteins can activate pro-apoptotic proteins and inhibit anti-apoptotic proteins, thereby fostering apoptosis. Extrinsic apoptotic pathway: after the death ligand binds to the death receptor, the death receptor interacts with the adapter protein FAS-associated protein with death domain (FADD) and recruits pro-caspase-8 to create the death-inducing signaling complex (DISC), and caspase-8 is activated, which can directly activate caspase-3/7, and can also cleave Bid into active tBID, thereby inducing MOMP-mediated apoptosis.
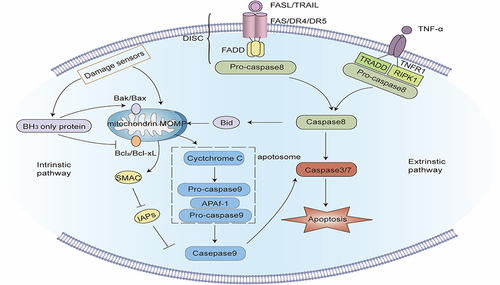
Figure 2 Signaling pathway of Autophagy. Macroautophagy: When a mechanistic target of rapamycin complex 1 (mTORC1) is blocked or 5’-AMP-activated protein kinase (AMPK) is activated, the Unc-51-like kinase (unc-51-like kinase, ULK) complex activates and phosphorylates the PI3K3 complex to start the nucleation phase of the autophagosome. Under the action of ATG4, ATG7, and ATG3, microtubule-linked protein1 light chain 3 (LC3) transforms into mature LC3-II, isolating ubiquitin-like substances into evolving autophagy. The binding of ATG5, ATG12, and ATG16 further stimulate autophagic membrane elongation. Finally, the autophagosome membrane seals to create mature autophagosomes, which merge with a lysosome to form autolysosomes, and the contents of the autophagosome are degraded by lysosomal enzymes. PINK1/Parkin-mediated mitophagy: When mitochondria are damaged, the protein kinase PINK1 is blocked from entering the inner mitochondrial membrane, but accrues in the outer mitochondrial membrane and is activated by autophosphorylation, and then recruits and activates Parkin. Parkin ubiquitinates several mitochondrial membranes substrates on the mitochondrial outer membrane. After these ubiquitinated substances are identified by ubiquitin-binding proteins such as p62, the damaged mitochondria are phagocytized and cleared by autophagosomes. Molecular chaperone-mediated autophagy: Molecular chaperone HSC70 particularly identifies and binds target proteins with KFERQ-like motifs, and transports these target proteins to lysosomes for degradation via the lysosomal membrane protein LAMP-2A.
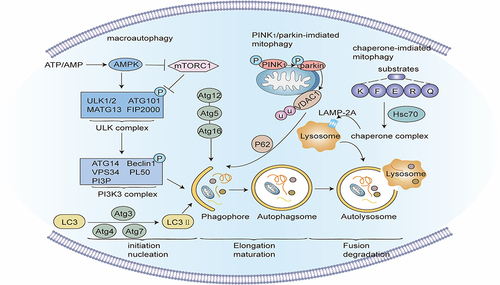
Figure 3 Signaling pathway of pyroptosis. Classic pathways of pyroptosis: Pathogen-linked molecular patterns (PAMPs) or damage-linked molecular patterns (DAMPs) activate the corresponding receptor proteins (NLRP3, NLRP1, NLRC4, AIM2 or Pyrin) and recruit ASC and pro-caspase-1 to form inflammasomes, and activate caspase-1. Caspase-1 hydrolyzes GSDMD to create GSDMD-C and GSDMD-N. GSDMD-N multimerizes and forms non-selective membrane pores on the cell membrane, elevating the permeability of the cell membrane, and resulting in cell swelling and rupture. Caspase-1 can also segment pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, and release them into the extracellular space through the above-mentioned membrane pores, inducing a strong inflammatory response. Non-canonical pathway of pyroptosis: Intracellular LPS directly binds and stimulates caspase-4/5/11. Activated caspase-4/5/11 can also cleave GSDMD, but cannot activate pro-IL-1β and pro-IL-18. In some cases, GSDME can also be hydrolyzed to GSDME-N by caspase-3/8, and the oligomerization of GSDME-N can also create plasma membrane pores. Notably, the hydrolysis of GSDMB by granzyme A and granzyme B can also form membrane pores, inducing pyroptosis.
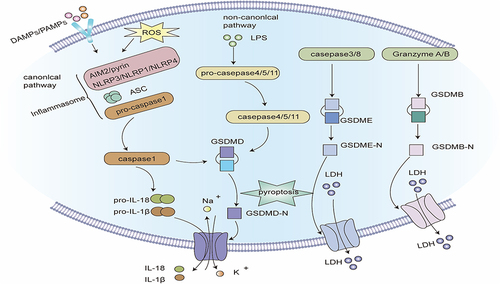
Figure 4 Signaling pathway of Ferroptosis. Extracellular Fe3+ is integrated with transferrin receptor 1 (TfR1) and then transported into the endosome, where it is lowered to Fe2+. Fe2+ is transported by divalent metal transporter-1 (divalent metal transporter 1, DMT1) and stored in the labile iron pool (LIP) in the cytoplasm. Nuclear receptor co-activator 4 (NCOA4) can transport ferritin to autophagosomes for lysosomal degradation and release Free iron. When iron metabolism is unbalanced, over-accumulated iron in cells will partake in lipid peroxidation via the Fenton reaction and iron-dependent enzymes such as lipoxygenases (LOXs). Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are major enzymes that control the percentage of polyunsaturated fatty acids (PUFA) in phospholipid membrane, which identifies the difficulty of lipid peroxidation. PUFA in membrane phospholipids is oxidized to phospholipid hydroperoxides (PLOOH) by the Fenton reaction or LOXs enzymatic reaction. GPX4 is a glutathione (GSH)-dependent enzyme that converts GSH into oxidized glutathione (GSSG) while reducing PLOOH to PL-OH, reducing oxidative stress, thereby negatively regulating ferroptosis. System Xc- consists of two subunits: the solute carrier family 3 member 2 (SLC3A2) and the solute carrier family 7 member 11 (SLC7A11). It is the common transport mode of cysteine, the main synthetic raw material of GSH. Inhibition of System Xc- will cause the depletion of GSH and the inactivation of GPX4, thus aggravating ferroptosis.
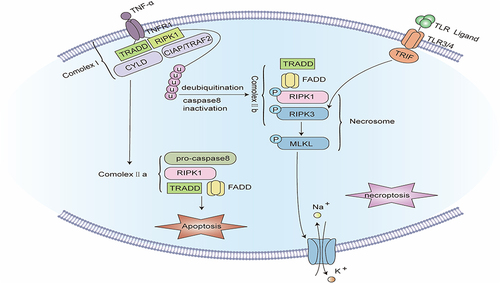
Figure 5 Signaling pathway of Necroptosis. The binding of TNF-α to tumor necrosis factor receptor 1 (TNFR1) promotes the assembly of complex I, including TNFR1, TNFR1-associated death domain (TRADD), receptor-interacting serine/threonine protein kinase 1 (RIPK1), TNFR-linked factor 2 (TRAF2), apoptosis protein (cIAP), and deubiquitinating enzymes such as CYLD. Complex I offers a platform for a series of subsequent ubiquitination and de-ubiquitination reactions, in which cIAP keeps RIPK1 ubiquitinated, while CYLD makes RIPK1 de-ubiquitinated. When caspase-8 is blocked, the ubiquitination of RIPK1 causes the formation of complex IIb. In complex IIb, RIPK1 is autophosphorylated, which subsequently recruits and stimulates RIPK3. PIPK3 further recruits and phosphorylates MLKL to create necrosomes. RIPK3 can also be independently stimulated by TIR-domain-containing adapter-inducing interferon-β (TRIF), an adapter protein of Toll-like receptors (TLRs). Eventually, phosphorylated MLKL oligomerizes and translocates to the cell membrane to develop plasma membrane pores, inducing plasma membrane rupture and cell disintegration.
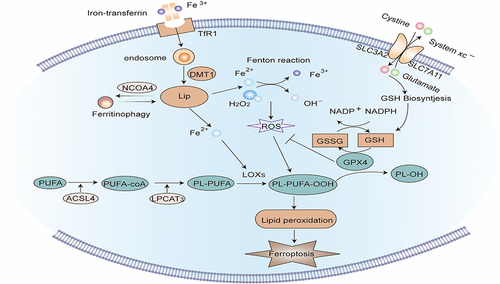
Table 1 Comparison of Apoptosis, Autophagy, Pyroptosis, Ferroptosis, and Necroptosis
Table 2 Therapeutic Strategies for Asthma Targeting Programmed Cell Death