
Figures & data
Figure 1 Two conceptual dichotomies among multiple forms of cancer cell death: ICD versus non-ICD, and PCD versus non-PCD. The multiple forms of cancer cell death include apoptosis, necroptosis, pyroptosis, and necrosis. The first three of these types of cell death are classified as PCD, and the last as non-PCD. PCD is defined as the death of a cell mediated by an active intracellular program, and is therefore also referred to as cellular suicide. Cell death can also be classified as ICD or non-ICD depending on the extent of DAMP induction or release from dying cells in response to chemoradiation. Representative DAMPs, as hallmarks of ICD, include CRT and HSP90 exposed on the outer surface of the cell membrane as well as ATP and HMGB1 released into the extracellular space. CRT, which promotes the engulfment of TAAs derived from cancer cells by DCs, is exposed only on cells undergoing immunogenic apoptosis, being absent on cells dying in an immunologically silent manner. Non-ICD can be accompanied by the release of immunosuppressive cytokines such as transforming growth factor–β (TGF-β) and interleukin 10 that are abundant in tumor tissue as well as by enrichment of regulatory T cells, both of which may promote tumor tolerance. The precise expression patterns of DAMPs and the balance between these two dichotomies in cell death types may determine the fate of the subsequent adaptive immune response namely, immune tolerance versus antitumor immunity.
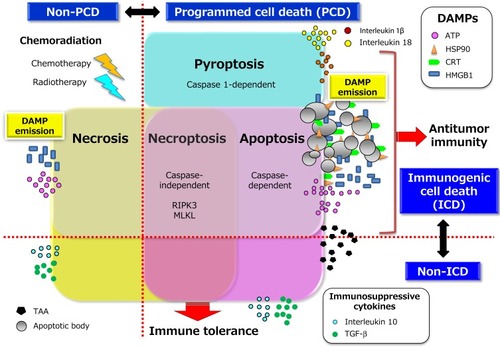
Figure 2 Chemoradiation-induced ICD and DAMP-mediated secondary antitumor immunity. The death of tumor cells induced by chemoradiation can be accompanied by the release or exposure of various DAMPs including ATP, HMGB1, and CRT. The interaction of these DAMP molecules with their cognate receptors including P2X7R, TLR4 or RAGE (receptor for advanced glycation end-products), and CD91, respectively can then facilitate both phagocytosis of primary and newly released TAAs by acting as an “eat me” signal as well as the maturation of APCs, particularly DCs. The mature DCs migrate to local tumor-draining lymph nodes to initiate priming of T cells mediated by the specific binding of antigenic peptide–MHC complexes on DCs to cognate TCRs. This binding elicits a signaling cascade from the TCR complex that ultimately triggers gene expression programs that induce the transition of the T cells from their resting state to a state of activation and proliferation. This transition also requires the presence of costimulatory signals elicited by the binding of the ligands CD80 or CD86 on APCs to their receptor CD28 on T cells as well as by that of the ligands OX40 and 4-1BB on APCs to their receptors on T cells. In general, the expression of costimulatory ligands is suppressed on APCs present in tumor tissue, but this suppression can be overcome by endogenous adjuvants such as certain DAMPs that activate pattern recognition receptors on APCs. Appropriate DAMP induction, acting through DC activation, may thus contribute to effective priming and generation of secondary CD4+ TH cell– or CD8+ CTL–mediated immunity directed toward primary TAAs and newly released TAAs including neoantigens (a phenomenon known as epitope spreading). TH1-skewing cytokines released from CD4+ TH1 cells amplify and sustain the expansion of CD8+ CTLs and their differentiation into effector and memory phenotypes. These multistep processes may eventually lead to the durable control of residual and micrometastatic tumor lesions by effector CTLs in a manner dependent on the induction of tumor cell death by directed exocytosis of cytolytic effectors such as granzymes. All abbreviations are defined in the text.
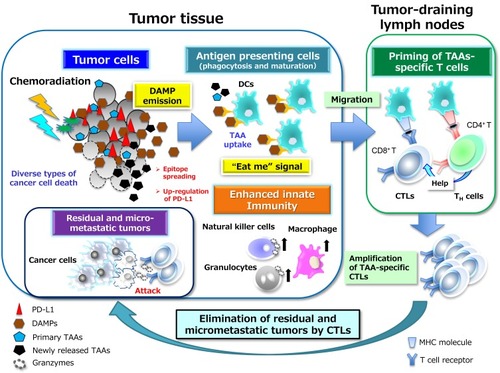
Figure 3 PD-1–PD-L1 interaction–dependent barrier to effector CTL activity in solid tumors. TAA-specific effector CTLs that can overcome chemokine mismatches and the aberrant vasculature of residual or metastatic tumor beds are able to attack cancer cells in a manner dependent on the interaction between peptide-MHC complexes and TCRs. The activated T cells secrete interferon-γ (IFN-γ), which induces the up-regulation of immunosuppressive mechanisms such as the expression of PD-L1 on cancer cells. PD-L1–mediated signaling via PD-1 displayed on effector CTLs renders them exhausted, anergic, or apoptotic, and thus no longer able to destroy cancer cells. However, inhibitors of PD-L1 such as durvalumab that interrupt the PD-1–PD-L1 interaction reinvigorate exhausted CTLs and restore them to the effector phase so that they are able to exert long-lasting antitumor effects in cancer patients. Immunosuppressive immune cells can also be attracted to tumor tissue via chemotaxis in response to chemokines induced by hypoxia. Inhibitory immune infiltrates include myeloid-derived suppressor cells (MDSCs), regulatory T cells, tumor-associated neutrophils (TANs), and tumor-associated macrophages (TAMs), all of which can produce immunosuppressive molecules such as interleukin 10, TGF-β, and prostaglandin E2 (PGE2). All abbreviations have been described previously.
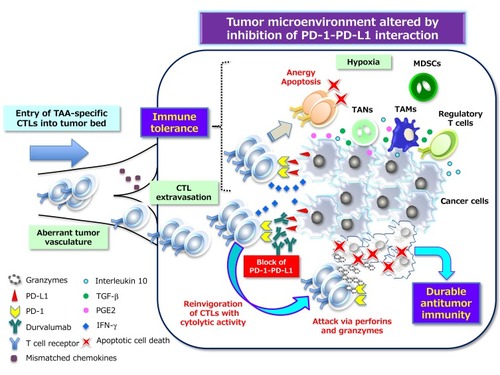
Table 1 Summary of Safety Findings for Japanese and Non-Japanese Subpopulations Treated with Durvalumab in the PACIFIC Study